您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-02-18 23:48
由于研发基础的不同,创新性药物和仿制药的研发模式和研发路径不同。创新性药物的研发,根据美国制药工业协会( PhRMA) 统计,平均每100 万个化合物经过前期筛选、临床前安全有效性评价以及临床安全有效性评价,仅有1 ~ 2 个产品可以获准上市,期间耗时约11 ~ 15 年,研发投入8 ~ 10 亿美元。创新性药物的研发路径是通过系统、规范、有针对性的临床前研究以及探索性临床研究,并进一步通过确证性临床研究确证药物的临床价值与安全性风险之间的关系即利益/风险比。不同研究阶段创新性药物的研究重心不同,药学研究的深度和广度是随着研发进展不断延伸的。由于创新性药物的研究是一个从未知、探索、确认“成药”可能性的过程,决定了其药学研究存在研究的不确定性和渐进性特点,主要体现在: ① 伴随临床研究进程分阶段推进。在临床前和早期临床研究阶段,药学研究主要是为以上研究提供质量相对稳定的研究用样品,随着临床研究的推进,药学研究则致力于稳定、重现、可工业化生产的生产工艺的确定以及完善质量控制体系的构建。② 结合临床研究/治疗需要以及放大生产需要进行变更,其剂型、规格、处方工艺、分析方法、质量标准等均可能在研究进程中发生变更。
由于创新性药物研究过程中药学变更的多样性以及药物的个性化特点,各药政管理当局均未对创新性药物研究过程中药学变更出台明晰的技术要求。本文旨在结合近些年创新性药物审评经验积累的基础上,提出一些关于创新性药物研究进程中药学变更的技术考虑。
1、变更的一般规律及评价核心
变更是药品开发研究中一个永恒的主题,根据多家国际大型制药企业创新性药物的研发经验,总结可知创新性药物研发过程中药学变更的常见原因为: ① 对于药物理化性质的进一步了解。② 为满足临床研究的需要。③ 为满足生产规模扩大的需要。创新性药物药学研究的变更主要发生在Ⅰ、Ⅱ期临床研究阶段,其原因在于越是后期变更可能对产品质量影响越大,变更的验证要求越高。
尽管变更是药品开发研究中一个永恒的主题,但是由于变更基础、变更原因和变更种类的不同,上市后变更和研发过程中的变更评价核心不同。对于上市后变更,需要强调的是变更后样品质量不得劣于变更前样品质量; 而对研究进程中的变更,需要关注的是变更前后样品质量的可衔接性; 通过评估可衔接性,重点考虑是否会对临床试验用药的安全性产生影响; 某些情况下,尚需考虑是否会对初步的有效性产生影响。例如,如果变更后原料药的粒度或晶型发生了改变,需要评估这种改变是否会影响原料药的溶解性,是否会影响制剂的溶出行为,是否会影响体内暴露量; 如果影响体内暴露量,则需进一步评估采用变更前样品在一定体内暴露范围内获得的安全有效性信息能否支持变更后样品在一定剂量下开展后续的临床研究。
2、变更的主要类型和技术考虑要点
2.1 原料药工艺变更
创新性药物研发过程中原料药工艺变更的主要原因为: ① 由于收率低、反应条件苛刻、成本高、起始物料可获得性差等原因,原开发的工艺不适合放大生产。② 由于新的工艺技术的应用。③ 可获得更适宜的起始物料; 如原工艺采用的起始物料无法商业化合法获得。
对于此类变更,研究中需重点关注: ① 变更后的工艺是否会引入新的有机溶剂、有毒试剂、催化剂等的使用。② 工艺变更是否会导致杂质谱的变化,如是否会导致基因毒性杂性的产生。③ 工艺变更是否会导致晶型改变,如重结晶工艺发生改变可能导致晶型改变。④ 由于杂质谱的变化是否需相应进行分析方法的修订和暂定限度的修订。⑤ 由于杂质谱、晶型等的变化是否会引起稳定性的改变。
2.2 盐基、晶型的变更
创新性药物研发过程中原料药盐基、晶型变更的主要原因为: ① 原采用的盐基或晶型批量生产比较困难。② 随着稳定性研究的深入,发现长期放置条件下原采用的盐基、晶型不稳定。③ 采用原盐基、晶型制备的制剂质量不理想。
对于此类变更,研究中需重点关注: ① 由于盐基、晶型的改变,是否会导致原料药相关理化性质的改变,如溶解性、吸湿性是否会改变。② 由于相关理化性质的改变,是否会导致体内暴露量的改变,如由于溶解性的改善,可能会导致体内暴露量的增加,进而引起安全性的担心。③ 盐基、晶型的改变是否会导致稳定性的变化,是否依然能支持临床研究期间的稳定性。
2.3 剂型、处方工艺变更
剂型、处方工艺变更是创新性药物研发过程中常见的一类变更。因在早期研究过程中,通常是结合药物性质,尽可能选择最简单易行且容易调整剂量的剂型。如口服固体制剂,通常选择胶囊剂,而且早期阶段倾向于直接用原料药填胶囊。随着临床研究的推进,由于研究用样品量的需求不断增大,并且获得了进一步的药物理化、生物学性质,则会调整至适合临床应用且能放大生产的剂型、处方工艺。归结起来,创新性药物研发过程中剂型、处方工艺变更的常见原因为: ① 满足临床研究的需要。② 满足放大生产的需要。
对于此类变更,研究中需重点关注: ① 处方的变更是否会引入特殊辅料的使用,该辅料是否会引发安全性担心。② 工艺的变更是否涉及无菌工艺改变,临床样品的无菌保证度是否会下降。③ 处方工艺的变更是否会导致杂质谱的改变。④ 剂型、处方工艺的变更是否会导致制剂特性的改变,如是否会引起制剂的溶出行为、制剂的含量均匀度等发生改变。⑤ 由于处方组成改变,是否需对原分析方法进行再验证或重建。⑥ 制剂特性的改变是否会引起生物利用度的改变。⑦ 剂型、处方工艺的变更是否会导致稳定性发生变化,能否支持临床研究期间的稳定性。
2.4 质量标准的变更
药品质量标准的建立是在信息积累的基础上不断完善的过程。早期研究中,质量标准的建立基于有限的研究数据,并且因为早期研究研究周期较短、受试者数量较少,故质量标准的制订也相对宽泛。随着临床研究的推进,积累了更多批次的研究数据,同时对于样品性质、生产过程也更加了解,质量标准的制订就更科学有据。归结起来,创新性药物研发过程中质量标准变更的常见原因为: ① 研究信息的不断积累为质量标准的科学制订提供了更充分的依据。② 由于关联变更导致质量标准的变更。
对于此类变更,研究中需重点关注: ① 如分析方法发生变更,应评估方法变更前后的检出能力是否一致或更好。② 如质控限度发生改变,如原料药的粒度限度变更,需分析粒度改变是否会导致制剂体内暴露量的改变、制剂含量均匀度的改变以及稳定性的改变。
2.5 产地的变更
创新性药物研发过程中产地变更的主要原因为: ① 早期系由合同工厂委托加工。如国内部分创新性药物是由科研单位开发,在并未选择好合作开发伙伴时,通常都是委托合同工厂加工制备早期临床样品。② 由于原生产厂生产能力有限,不能满足生产规模扩大的需要。
对于此类变更,研究中需重点关注以下内容:① 变更后产地的GMP 状态。根据《药品注册管理办法》,临床样品的制备须在符合GMP 要求的车间进行。② 变更产地过程中技术转移的有效性。具体表现在: 变更产地后处方、工艺的一致性,生产设备原理的一致性,分析方法的一致性,变更产地前后制备样品相关质量特性的比较( 包括体外、体内比较) ,变更产地后样品的稳定性。
2.6 其他
在创新性药物研发过程中可能发生的其他药学变更包括: 规格变更,贮存条件和包装系统的变更。通常规格变更是伴随临床研究的推进,随着临床推荐剂量的确定进行变更,以更方便临床用药。该类变更通常伴随着剂型/处方工艺的改变,故研究思路同“3.3”项下所述。
贮存条件和包装容器/系统的变更通常是根据长期稳定性研究数据的积累进行相应调整。在药物的早期开发阶段,由于仅可获得有限的药物稳定性信息,通常会选择较为保守的贮存条件,比如对于相对不稳定的原料药采用- 20 ℃保存条件。随着研究推进,在比较了- 20 ℃、2 ~ 8 ℃、25 ℃下的长期稳定性情况后,选择适宜的贮存条件。
3、案例分析
下面分别以原料药工艺变更和制剂剂型、处方工艺变更的具体实例对以上研究思路做进一步阐述。
3.1 原料药工艺变更
3.1.1 背景信息
某原料药,含有一个手性中心,水溶性差,存在两种晶型( 晶型A 和晶型B) ,临床制备为胶囊剂,规格0. 5 mg。
3.1.2 变更历史
本品在进行Ⅰ期临床时,采用拆分工艺( 下文表述为工艺A) ,生产批量可达到1.8 ~ 2.2 kg。进入Ⅱ期临床,由于临床样品需求量大,需扩大生产规模,原拆分工艺无法进一步扩大生产规模,故研究开发了不对称合成工艺( 下文表述为工艺B) ,生产批量可达到5.4 kg。
3.1.3 研究工作
3.1.3.1 杂质谱的分析及杂质控制从安全性角度考虑,研究中首先需要验证的是由于工艺路线的改变是否会导致杂质谱的变化,具体杂质谱对比分析见表1。
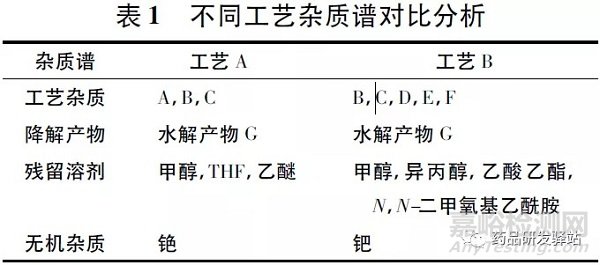
从表1 可以看出,相比于工艺A,工艺B 获得样品中增加了3 个新的工艺杂质,引入了3 种新的有机溶剂,同时使用了金属催化剂钯。
针对杂质谱的变化,研究者重新建立了有关物质、残留溶剂测定方法,进行了相关验证,并参照ICH 指导原则设定新增杂质的限度; 因工艺B 中采用钯做催化剂,考虑到钯为毒性较强的Ⅰ类重金属,采用ICP-MS 法对其进行残留测定,并参照EMA 相关指导原则设定残留限度。
3.1.3.2 关键理化性质的比较因该原料药存在多种晶型,故研究中重点考察了工艺B 获得样品晶型是否同工艺A 获得样品晶型一致。通过DSC、粉末-X 衍射等分析确认晶型一致。
3.1.3.3 变更后稳定性考察由于工艺路线改变导致了杂质谱的变化,并且Ⅱ期临床研究的样本量大于Ⅰ期临床、临床研究周期长于Ⅰ期临床,为支持Ⅱ期临床研究期间的稳定性,采用两批工艺B 生产原料药( 批量为5.4 kg) 以及两批工艺B 生产原料药制备的制剂进行了6 个月的加速和12 个月的长期稳定性考察,结果提示样品稳定。
3.2 剂型、处方工艺变更
3.2.1 背景信息
某原料药,属于BCS 分类Ⅲ的化合物,溶解性随着pH 的增加下降,主要降解途径为水解,临床设定剂型为口服固体制剂。
3.2.2 变更历史
本品在Ⅰ期临床研究中制备为胶囊剂,采用原料药粉末直接装胶囊,规格为15,200 mg。Ⅱ期临床研究中,为了改善粉体的流动性并提高临床使用规格,采用干法制粒( 20% 载药量) 后装胶囊,规格为100,200 mg。Ⅱ期临床研究确定给药剂量为800mg bid,Ⅲ期临床研究中,为了提高规格、改善可压性和外观,采用湿法制粒( 40% 载药量) 后装胶囊,规格为200, 400 mg。Ⅱ、Ⅲ期临床研究阶段处方对比见表2。
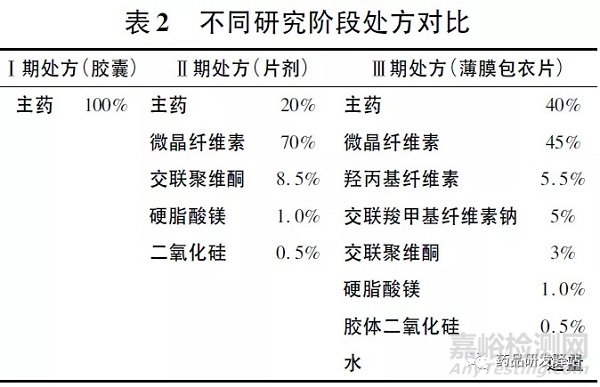
3.2.3 研究验证工作
3.2.3.1 溶出度对比考察相比于Ⅱ期临床研究样品,Ⅲ期临床研究样品的处方工艺发生了较大变化,故首先需要考察如上处方工艺的变化是否会导致样品溶出特性的改变。对于变更前后的样品在pH 1.2,pH 4.5,pH 6.8( 其中加入1% 吐温80) 3种介质中进行了溶出行为的比较,结果显示溶出行为基本一致。
3.2.3.2 BE 研究考虑到处方工艺变更较大,且将进入关键性的Ⅲ期临床,在体外溶出对比一致的前提下,进行了变更前后样品的BE 研究,以保证体内暴露量的可衔接性。
3.2.3.3 分析方法的验证因采用辅料种类不同,故需验证原建立方法的适用性,主要考察了有关物质、含量测定以及溶出度方法,重点验证新增加辅料是否会干扰可能的降解产物以及主成分的测定。
3.2.3.4 杂质分析由于处方组成和生产工艺不同,可能导致杂质种类和杂质量的不同。对于变更前后样品进行了杂质对比分析,重点考察了水解产物的变化,结果显示湿法制粒工艺并未导致水解产物的增加。
3.2.3.5 稳定性考察由于制剂的处方工艺均进行了调整,故采用修订后处方工艺生产的三批样品重新进行了6 个月的加速和6 个月的稳定性考察,并承诺继续进行后续的稳定性研究。
5、结语
由于创新性药物药学研究具有不确定性和渐进性的特点,决定了其研究过程中必然存在变更,但是变更要有因有据,应明晰变更的目的、需变更的内容并开展相关研究验证变更是否达到预期的目的。对于创新性药物研发过程中药学变更的管理,研究者需关注以下内容: ① 依据变更的种类、变更的程度以及变更发生的阶段开展相应的研究验证工作,自我评估变更的影响,分析变更前后样品质量的可衔接性。② 制订变更控制程序,以保证研究的规范性和科学性。③ 系统记录变更历史,以保证研发信息的全面性。

来源:Internet


