摘要:该文是在国家鼓励高质量推动中药产品国际注册的大背景下,针对美国植物药注册申请的药学要求开展的研究。研究以现行版《美国植物药开发工业指南》为基础,以我国在FDA 注册申报的复方丹参滴丸和连花清瘟胶囊2个产品以及USFDA 已批准的4 个植物药处方药产品为案例,基于公开信息剖析其中的药学研究内容,探究美国植物药注册申请的药学研究关注点和难点,以期为我国中药1.2 类提取物及其制剂、天然药物的研发和我国中药在FDA 以植物药处方药注册申请提供参考和借鉴。
美国植物药由植物组成,包括植物材料、藻类、大型真菌,或者上述物质的组合。剂型可以是溶液剂(例如茶剂)、粉剂、片剂、胶囊、糖浆、外用制剂或注射剂。发酵类、高纯度、化学修饰的植物原材料等不属于植物药范畴。目前美国已批准4 个满足植物药指南定义的新植物药处方药[1]。
1 植物药的特点及其所面临的科学和注册方面的挑战
植物药源自天然,由复杂的混合物组成,活性成份不明确,多种化学成分可能对生理或药理作用有贡献,其化学成分及生物活性,在质量控制方面通常无法得到很好的表征。此外,植物药还存在较大的批间变异性,这种变异性比化学药的批间差异更为显著。化学药的常规质量控制方法对于植物药来说也是不充分的,植物药通常需采用多种方法进行联合检测。相比于化学药,植物药通常具有先前的人用经验,可以为早期临床试验安全性提供一些提示,但在后期临床试验中,需评价批间差异对植物药疗效的影响。
在管理方面,植物药与其他药品一样,均需符合同样的法律标准[2]。但是植物药提供的某些资料可以与新化学实体(NCE)不同,主要包括以下方面,见表1。
由于植物药的上述特点,使得对植物药来源、生产工艺步骤、标准化和处方过程等方面的理解,对植物药制剂的特性鉴定和一致性评价都至关重要。此外,植物药的生物作用机制可能未知或由于过于复杂而无法明确获知。当无法进行药理学和药代动力学试验时,可以豁免,但在缺乏此类数据的情况下,可能更难确定植物药的“洗脱”期,或预测和研究“药物-药物”相互作用。植物药在临床应用中的很多处理方式与复杂的生物制剂更为相似,美国食品药品监督管理局(USFDA)采用的处理方法更类似于生物制剂。由于植物药的特性无法全面表征,因此,植物药通常通过过程控制和验证来定义,即“过程即产品,产品即过程”。不过,这种复杂性反而使得植物药在没有专利保护的情况下也能得到知识产权保护。在审评植物药申请时,USFDA 会考虑这些因素,并根据“个案处理”的原则向申办方提供建议[3]。对于以植物原材料为起始物料的药品,其生产首先要确保植物原材料(botanical rawmaterial,BRM)的鉴定,此外,应该考虑BRM 的可及性及持续供应,上述内容都属于良好种植和采收管理规范(GACP)的范畴。USFDA 通常把植物原料药(botanical drug substance,BDS)整体作为活性成分,BDS 的表征较难,可以通过生物标志物和化学指纹进行表征;每个工艺步骤均应进行详细的描述;全过程的控制有助于确保临床上所用剂量的可重复性以及批间一致性。
2 美国植物药的药学研究要求
基于植物药的上述特点和注册方面的挑战,USFDA 专门起草了《美国植物药开发工业指南》用于指导植物药的研发和注册申报[4]。本文将集中在其中的药学内容进行概括、梳理和探究。
2.1 不同研究阶段的一些总体要求和建议
2.1.1 在通用技术要求方面,植物药的与其他药品一致
植物药是药品的一种,在质量控制和临床试验设计方面与其他药品相比有其特殊性,但是同时它也需要遵循其他药品的一些通用要求。例如:植物药制剂的新药申请需要采用电子通用技术文档(eCTD)格式递交;稳定性试验参考ICHQ1A(R2)的科学原理进行稳定性试验设计;植物原料药可以作为临床试验申请(IND)、上市许可申请(NDA)资料的一部分递交,也可以以药物主文件(DMF)的形式递交。
2.1.2 注册资料的提交要求
植物药的临床开发采取循序渐进的方式,以便于采用早期(I期、II期)的临床试验结果支持后期(III 期)的临床试验设计。早期临床试验需提交的资料取决于以下因素:先前的人用经验和既往临床试验的研究程度、药物的已知或可能风险以及药物的开发阶段。对大多数植物药来说,早期临床试验阶段可能无需提供详细的药学(CMC)资料(例如:原料的全面表征数据),在III期临床试验启动前需要提交较完善的药学研究资料。
2.1.3 留样建议
在早期临床试验时,临床试验用样品应留样,以备USFDA 和/或申办方未来的检验。临床样品所用的植物原材料和原料药生产商的每一批样品均应留样;在期临床试验时,应保留充足的、不同批次的植物原材料和原料药,以备后续的化学表征和/或药理/毒理试验。
2.1.4 稳定性试验要求
在早期临床试验时,原料药需开发稳定性指示性的分析方法,制剂稳定性数据应至少支持临床试验期间的使用。稳定性试验贯穿药品上市的全过程,并最终支持拟定的有效期。在新药申请时,稳定性试验方案中应包含生物测定方法,申请人应开发、验证和使用具有稳定性指示性的分析方法和/或生物测定方法,以监测植物原料药和制剂的稳定性。申请人还应进行影响因素稳定性试验,以识别原料药和制剂的降解产物,评估潜在的毒性,并对这些降解产物进行充分控制。
2.2 不同研究阶段,针对可能的变化,需要开展的桥接研究
2.2.1 早期临床试验
由于植物原材料在种植和采收过程中可能的变化和/或工艺优化而导致的生产工艺条件的变化,进而导致不同开发阶段所用的植物原料药可能会在某些特性上有所不同(例如化学组成),需要通过桥接研究来说明这些变化对质量产生的影响,桥接研究的类型需要与新药办公室(OND)的审评部门沟通。
2.2.2 III期临床试验
在III期临床试验前,原料药和制剂的药学开发应足够深入,以确保在III期临床试验过程中不对植物原材料和生产工艺进行重大变更,从而确保临床试验用原料药与拟用于上市的原料药一致。
如果 IND 阶段提供了或引用了早期开发阶段的非临床和/或临床数据,申办方应提供IND 所用植物药制剂与引用研究中所用植物药制剂间的比较研究(例如:原料药中活性成分或化学成分的特性鉴定和定量测定、制剂的组成和配方等)。同样,在开发过程中,当植物原材料、原料药或制剂的来源和生产工艺发生变更时,申请人应对变更前后进行对比,来源和/或生产工艺看似微小的变更,也可能导致临床疗效的显著性差异,进而会导致早期的药理、非临床和临床数据不可用。如果无法确定不同批次原料药是否相似,应提供桥接研究(例如:原料药中活性或化学成分的化学鉴定和定量分析、生物测定、和/或其他非临床研究)来证明不同开发阶段原料药的充分相似性,进而判定先前的非临床研究和临床试验结果是否可用。
处方的变更若导致先前开展的药理或毒理研究无法适用于新的处方,可能需要提交非临床桥接研究。申办方应就拟定的处方变更与审评部门进行咨询,确定变更是否需要桥接研究或其他研究。值得注意的是,如果在临床试验开发阶段,原料药和制剂发生变更,可能需要提供非临床桥接研究。
2.2.3 新药申请阶段
后期临床试验用的原材料的采收、加工方法应与早期所用方法相同。临床开发过程所用的原材料的采收、加工方法发生变更的,可能会改变原料药、制剂的化学组成,需要提交桥接研究以证明先前的临床试验结果仍然适用。
2.2.4 上市后阶段
植物药制剂变更所产生的影响可能不易识别,有必要开展额外的研究(例如:生物测定和/或其他体内桥接研究),评估效力和活性。USFDA 将根据变化的性质和程度以及混合物中活性成分或化学成分表征的程度等具体情况,要求申办方提供相应的研究数据。
2.3 生物测定法
生物测定法是一种测定植物药效力和活性的重要方法。首选与药物已知或预期作用机制相关的生物测定法。某些情况下,可以考虑采用相关性较低的检测方法进行评估。由于生物测定结果的变异性较大,所以应测定相对于对照品或对照物质的批效力和活性,生物测定结果应以对照品或对照物质校正的活性单位来表示。申请人应开展系统适用性考察和质量控制研究,以确保检验方法的重复性和可预测性。如果植物药的活性成分未知或无法定量,在III期临床试验开始前,应开发生物测定方法,以评价原料药相对于对照物质的批效力和活性;在提交新药申请时,申请人应对生物测定法进行适当的验证,验证指标至少应包括准确性、精密度、专属性、线性和范围。如果质量控制需要采用生物测定法,质量平衡应与生物测定法联合进行评价。
稳定性试验方案中应该包含生物测定法,变更研究时生物测定结果也是一项关键的对比指标。
2.4 质量平衡
在III期临床试验开始前,应开发其他(例如脂质或蛋白质)定量方法,以测定对原料药质量平衡有贡献的其他类成份。
在新药申请阶段,植物药开发过程中积累的经验和数据应为制定临床相关标准奠定基础,分析方法应通过适当验证。此外,当联合使用多种方法时,所有分析方法所得到的整体数据,应可以证明检验样品中不同类成分间和同一类成份内(如果恰当)的质量平衡。分析方法应足够敏感,以检测多批次关键质量属性之间的差异。如果可能,原料药应包括以下检验内容:各已知化学成分的重量和同类成分的总量、各未知峰面积百分比、各未知可量化峰的相对保留时间(RRT)、总脂和各脂肪酸的重量、总氨基酸和各氨基酸的重量、简单碳水化合物总重量、复杂碳水化合物总重量、总维生素和各维生素重量、灰分含量等。
2.5 确保植物药疗效一致性需要提供的证据
植物药制剂生产商面临的重大挑战之一是确保不同上市制剂批次(尽管有变异性)的疗效与III期临床试验用样品疗效的一致性。由于植物药制剂天然的变异性,仅靠化学检测可能不足以控制其质量,无法保障疗效的一致性。植物药制剂的质量控制应从以下3 个方面考虑:①植物原材料的控制,②化学检测的质量控制和生产控制,③生物测定和临床数据。申请人应对上述3 个质量控制方面进行综合评估,以确定所需的数据量,例如,①和/或③(生物测定)所需的数据量可能取决于②化学检测对植物混合物中成分表征的程度,从而证明上市后的各制剂批次与上市前临床试验用样品疗效一致。NDA 申请时,这部分资料放在模块2.3.P.2(药学开发)项下植物药特有的章节“治疗一致性保证”。
尽管原料药控制和其他 CMC 控制措施有助于产品的特性鉴定,保证质量,但在某些情况下,可能需要提供有关质量指标与药理学活性或临床疗效之间的相关性,以确保原材料和原料药的变化不会影响制剂的疗效一致性。临床数据包括剂量—效应数据和多批次制剂的数据。通过剂量—效应数据,若发现临床疗效对剂量不敏感(但仍优于安慰剂对照组),则有理由推测,质量标准范围内的变化可能不会影响药品的疗效一致性;多批次制剂的临床数据,是指临床试验中将受试者随机分配到不同的制剂批次,用于评价疗效与制剂批次的相互作用。若相互作用不明显,说明质量标准范围内药物批次的变化不会影响疗效[5]。
2.6 不同研究阶段对植物原材料(BRM)、植物原料药(BDS)和植物药制剂(BDP)的具体要求
2.6.1 早期临床试验阶段
2.6.1.1 BRM
应该提供先前人用经验中有关BRM 的以下信息:国际双名法惯例命名、异名、种(亚种、变种和变型)和栽培变种、科名、药用部位;关于化学组成,应识别已知活性成分群(确定为特定化合物群或化学类别)和未知活性成分群的化学成分(例如,作为鉴别和质量控制的特征)。
支持 IND 所需的CMC 信息量取决于很多因素,包括植物药的上市历史和研发阶段,在美国境外上市的植物药人用经验数据要求取决于境外生产该植物药的控制措施和质量控制的完整性。
对 BRM 的控制包括通过感官、外观和显微方法鉴别植物基原、药用部位等,以及通过与凭证标本(即对照标本)对比进行的鉴别研究,还可以通过遗传分类学的方法进行鉴别(DNA 条形码技术)。如果所用物种包含多个变种时,应明确每个变种。植物原材料和原料药生产商的每一批次样品均应留样,并在适当的条件下保存植物及其药用部位或其他植物材料。必要时,可以用这些样品来鉴别真伪。
需要提供的资料包括:植物种及其药用部位的鉴定、真品凭证、物种是否属于濒危物种及是否生长在濒临灭绝或受到威胁的产地中。
应提供每个种植者和/或供应商(如果有)的名称和地址,植物种(变种和栽培变种)的外观和特征,以及植物鉴定(外观和显微)信息,产地(全球定位系统GPS 坐标)、生长条件、采收时植物的生长阶段、采收时间/季节,和后续的加工处理(例如:清洗、干燥、研磨工序);杂质控制(即:无机和有机污染物,如土壤、昆虫和藻类/真菌);保存;搬运、运输和贮藏条件;元素杂质检测;微生物限度;残留农药检测;和外源性毒素检测(例如黄曲霉毒素)、外来物质与混伪品等。
2.6.1.2 BDS
应提供原料药的定性描述、定量描述、生产商、生产工艺描述、质量控制信息等,具体内容如下。
原料药的定性描述包括名称、外观、活性成分、理化性质、生物活性、以及先前临床中用于制备原料药的各个植物原材料。如果活性成份、生物活性或先前临床使用情况未知,应该在申请时阐明。对于多种植物组成的原料药,申请时应说明原料药的各植物原材料单独加工成原料药后再混合还是混合后再加工。
原料药的定量描述应包括:植物原材料的规格(一般以加工后的植物原材料的绝对干重计);应该提供批量以及相对于植物原材料的得率;当活性成分或其他化学成份已知、可测时,应该明确其在植物原材料中的含量;对于多种植物组成的原料药,应该阐述每个加工后植物原料药或加工前植物原材料(若适用)的相对比例。
生产工艺描述。应包括每一个工序药材投料量、溶剂、提取和/或干燥的温度和时间及生产过程控制;应标明加工收率,以原药材的量相对于提取物的量来表示;如果用多种植物原材料混合生产含有多种植物的一个原料药,应提供每个药材的投料量及其加入顺序、混合、研磨、和/或提取等信息;如果多植物原料药是由两种或两种以上单独制备的植物原料药共同组成,应对每个原料药的工艺进行单独描述。
原料药的质量控制。对每批原料药进行质量控制,包括所用的分析方法、检验结果及拟定的可接受标准。质量控制检验项目至少应该包括:外观、以干重计的规格(相当于植物原材料的量)、活性成份(如果已知)或化学成份的鉴别和含量测定。一般来说,申办方应该采用可用的分析技术解决分析方法的分离度问题。应采用多种方法互相补充确保对成份进行充分的化学定性和定量表征。若采用多种植物原材料混合生产一个原料药,无法对每个活性成分或化学成分进行定量测定时,可以建立一种联合方法对多个活性或化学成分群进行测定。当多个活性成分或化学成分已知时,应对其进行化学表征,并对其相对含量进行定义。此外,还应对农药残留(经常使用的农药)、无机杂质、残留溶剂和放射性污染(如果有的话)、微生物限度、外源性毒素(如黄曲霉毒素)等进行检验,以及开展稳定性试验、生物测定法以及质量平衡等研究。
2.6.1.3 BDP
植物药制剂应提供以下资料:①制剂的定性描述,如剂型、给药途径、组成制剂的物料名称和功能(例如:植物原料药、其他原料药和辅料),对于植物原料药与其他原料药(例如,高纯度的、生物技术的,或其他天然来源的药物原料)的复方,应予以说明;②制剂的组成或定量描述,以单位剂量和批处方量来表示;③制剂生产商的检验报告或制剂生产商授权USFDA 对生产商之前提交的资料或DMF 中的相关CMC 信息进行交叉引用。如果国外上市产品没有这部分信息,则申办方应对制剂进行质量检验。除了这些检验外,应开展无机杂质分析和动物安全性试验(如适用),申办方应当在IND 时提供检验方法和结果。留样建议以及稳定性数据要求在前面已有阐述。
特殊情况下,如原料药的活性成分已知,且药材中这些活性成分的浓度有较大的、天然的变化(例如:因生长条件随时间推移而产生的难以控制的变化),允许植物药制剂增加单体活性成分的含量以满足基准原料药的质量标准,以及疗效的批间一致性。但加入的活性成分目标含量不应超过天然含量。关于加入活性成分含量的恰当与否以及确定基准原料药标准的过程等相关事宜,申办方应事先向USFDA 咨询。
安慰剂。必要时,可在安慰剂中使用某些植物材料来掩盖活性药物,但这类植物材料不应有明确的药理活性,如有活性,将使临床试验数据难以解释。对于一些植物药来说,很难制备出一种在味道、气味和外观上与活性药物相同的安慰剂。如果研究者和受试者区分不出活性药物与安慰剂,并且可以保证临床试验的盲法,那么即便安慰剂与试验药物间有细微差别也是可以接受的。就该类安慰剂的使用,鼓励申请人向OND 相应的审评部门咨询。
2.6.2 III期临床试验阶段
植物药深入而持续的表征可以确保原料药的质量,进而保障临床数据的有效性和可靠性。
不同批次植物原料药间会有批间差异(例如:化学成分的变化),申办方应提供证据证明原料药的批间差异不会对疗效产生明显影响。其中的一种方法是在III期临床试验中使用多批次的植物药制剂(即每批制剂采用不同批次的植物原材料生产),以研究不同批次制剂间的临床疗效。这些研究将有利于申办方更好地了解与临床疗效相关的变化因素,以及在多大范围内的变异仍可保证植物药制剂的质量、有效性和安全性。这种深入的研究将有助于制定临床相关质量标准的可接受限度。
2.6.2.1 BRM
为评价质量和疗效的一致性,选择具有代表性的BRM 批次生产临床试验用原料药,进而生产多批期临床试验用制剂。申请人应建立3 个或更多的种植基地或农场等大种植区,这些种植区应选择在各BRM 的代表性区域中,并按照GACP 的原则进行种植。这将有助于在NDA 批准后减少药材资源不足的风险。
申请人应提供药材表征的进一步研究信息(例如:通过光谱或色谱法对每个药材进行化学鉴定,必要时用DNA 指纹图谱法对每个药材进行真伪鉴定),更新药材供应商所用的质量控制方法、分析方法以及拟定的质量标准,以便为NDA 资料递交做准备。
2.6.2.2 BDS 及BDP
申请人应建立相关质量标准(含暂定的可接受标准),并根据期临床试验结果在NDA 申报时确定最终的质量标准。
2.6.3 新药申请阶段
鉴于植物药制剂的独特性和特殊考虑,新药申请前会议尤其重要。一般来说,植物药制剂的NDA 资料提交要求与其他药物相同。
在 IND 阶段提交的有关制剂描述和先前人用经验资料的所有信息都应该在NDA 中提交,若有最新的人用经验资料(例如:基于已在国外销售的类似产品),应进行更新。
质量控制。由于原料药的活性成分可能未被识别,质量控制的技术挑战是确定植物药的特性并确保其疗效的一致性。植物药制剂质量控制的整体证据链条法(totality-of-the-evidence approach)应延伸到对原材料可能采取的额外的控制措施,如生物测定和/或多批次临床试验中BRM 质量差异对临床结局的影响。对植物药的鉴定不仅在于对混合物中化学成份的特性鉴定,还要依赖化学检测的质量控制和生产控制、生物测定、临床数据等。
2.6.3.1 BRM
植物制剂的质量控制应从 BRM 开始,并在NDA 中进行阐述。应明确药用植物(如采用形态学、宏观和微观分析、化学分析等方法鉴定真伪)、种植规范(如生长、采收时间、贮存条件)、地理位置,以及采收和加工方法的具体信息。申请人在提交NDA 时,应建立了GACP 基地,并汇总每种BRM 的相关规程。应参照世界卫生组织(WHO)、欧洲药品管理局(EMA)或BRM 种植区监管机构制定的GACP 通则。当植物的分类较复杂,或存在BRM 鉴定问题时,可以采用DNA 指纹图谱鉴定。例如,如果多个相关植物药种属用于生产某特定药材时,DNA 指纹鉴别方法较其他鉴别方法专属性更强。此外,申请人应描述减少植物污染、退化和变异的方法,早期和后期临床试验所用的植物原材料应该使用同样的方法。在临床开发过程中,这些采收加工方法上的变化可能会改变原料药,进而导致植物药制剂的化学组成的变化,需要提交桥接研究以证实先前临床试验结果的可靠性。
2.6.3.2 BDS 和BDP
①特性鉴定。表征植物药的所有化学成分存在一定困难,不仅体现在对混合物的化学特性进行表征,还包括对植物原材料的控制、生产控制、生物测定、临床数据等。即便如此,申请人还应利用现有技术和新技术开发正交分析法,对植物药中活性成份或化学成分进行充分定性和定量。当活性成分未知、植物混合物不能被完全表征时,申请人可以选择一种化学成分特征谱(这种特征谱对BRM 质量和/或原料药、制剂生产条件变化较敏感)进行特性鉴定。②化学特征。在NDA 阶段应充分阐述用于植物药化学成份表征的多种分析方法。NDA 阶段应包含用于定性和定量表征的活性成份或化学成分的所有化学检测方法,也包括提供数据说明质量平衡。③生产工艺。申请人应提供用于商业化生产原料药和制剂生产场地的所有信息。应避免变更原料药的生产场地,特别是临床开发后期。NDA 应提供完整的生产信息,包括所用的生产设备、中间控制和检验。④其他。还应提供生物测定法、质量平衡等质量控制资料以及稳定性试验资料。
2.6.4 cGMP 要求
鉴于植物药活性成分的异质性和不确定性,对植物原材料的控制,包括贮存条件和加工方法,都极其重要。申请人应在申请中提供充分的起始物料质量信息,而不仅仅依靠对终产品的质量控制。植物原料药的生产应符合cGMPs。在某些情况下,可能需要同时符合GACP 和cGMPs,以覆盖药材的种植、采收、加工和贮存的各个方面。申请人应指定原料药生产的起点并论证其确定依据。
2.7 上市后考虑
鉴于 BRM 稳定来源和原料药生产工艺一致性的重要性,制剂批准后若对产地、种植和采收规范、和/或加工方法等进行变更,均须仔细评估,以确保变更前后所生产的制剂在药理和/或疗效上的充分相似性。对于植物药制剂,变更的影响可能不易评价,必要时需开展额外研究(例如采用生物测定和/或其他体内桥接研究)来评估其效力和活性。针对变更所需要的研究数据,FDA 会具体问题具体分析,例如会考虑变更的性质和程度以及混合物中活性成分或化学成分的变化程度。
3 我国中药在USFDA 申报品种的药学研究情况
根据文献报道,我国已有一些中药品种在美国获批了IND 申请,主要开展了II期临床试验,进入III期的寥寥无几。基于可查询到的公开文献信息,以2 个品种作为案例,介绍其药学研究情况:
3.1 复方丹参滴丸(T89)
复方丹参滴丸于 1997 年以天然复方混合制剂的形式首次申报USFDA 的IND 申请,直接进入新药II~II期临床试验,2006 年再次通过USFDA 的IND 申请,2008 年在美国启动期临床试验,2010年完成期临床试验,2016 年完成期临床试验。1997 年获批开展临床试验后,对复方丹参滴丸质量标准进行深入研究,例如增加含量测定、指纹图谱、开展含量均匀度、重金属和农残检查等,制定原药材、半成品、成品的质量标准或质量控制方法[6-14];生产全程符合GMP 规范,药材种植符合GAP规范要求[15-29],2003 年丹参种植基地通过认证[30-35]。完成了FDA II 期临床样品溶出度、溶解度、指标成分含量测定方法及鉴别方法、溶剂残留、安全指标成分等一系列检验方法的研究工作[36-40],为药效物质基础研究及指导工艺生产奠定基础,顺利保证了期临床试验的成功[41-43]。
II期临床样品和III 期临床样品均在符合GMP 条件下生产。期临床试验前天士力全面实施了CMC 工艺再提升、质量标准再完善[44-54],创新了制备工艺,创造了新技术和新装备[55-56],优化了提取与制剂生产线、实施了厂房设备设施与工艺方法的全面再验证,提升了生产过程中的质量控制手段[57-58]。CMC 确定了以提取物作为活性整体,并参照化学原料药方式设定提取物质量标准,通过实施提取物批次搭配(混合)投料提高指标成分的控制精度;USFDA 基于我国和EMA 相关指导原则要求,接受了含量均匀度的测定方法和稳定性指标的显著性差异问题;提取物标准除了强调常规的安全性检查项目外,还根据植物来源和药效作用,增加侧柏酮、硝酸盐等检测项目;质量一致性评价方面,药材基源需鉴定至种及变种、亚种,强调产地、良好种植采收管理,药材处理、加工及储存要遵循GMP 管理。根据工艺质量要求,强调批内均匀性、质量稳定性。对提取物及制剂,要严格执行USFDA 的cGMP 要求,在厂房、设备设施、工艺质量设计及管理方面建立全面质量管理与风险评估。含量控制指标的建立与评价可结合药理药效、临床样品设计及临床试验的结果逐步提升与优化。此外,天士力集团结合T89 的作用机理、药效特点探索开发了基于斑马鱼急性心肌缺血模型的生物效价方法;关于质量平衡问题,USFDA 建议提供化学标志物占提取物的重量百分比并对提取物中其他未明确的组分加以讨论,要求在上市前提取物和制剂的质量标准中,设定大类组分的标准用于阐明质量平衡。USFDA 一方面认可提取物活性整体的质量控制复杂,另一方面又希望通过化学组分的质量平衡、生物效价等研究来寻找到提取物活性整体的生物等效性评价方法,以方便产品生产过程质量控制评价以及上市后的变更评价。天士力在II 期临床试验后,全面实施了符合USFDA 标准的cGMP 的质量管理。CMC 研究是基于质量风险管理,提倡质量源于设计、依靠技术创新、强化过程控制、实施全面验证、持续完善标准的系统工程[59-60]。
3.2 连花清瘟胶囊(KT07)
2013 年10 月以岭药业提交连花清瘟胶囊pre-IND 沟通交流,针对USFDA 正式回复,制定工作方案,包括药材GAP 基地、药品的CMC 到临床、药理、药学、生产等环节。2015 年12 月获批在美国开展II期临床试验。
①以岭制剂工艺优化开展了以下桥接研究:陶瓷膜技术代替原醇沉除杂工艺(以膜通量、水提液出膏率以及有效成分和指纹图谱为指标);真空带式干燥技术代替喷雾干燥(对影响产品质量的关键环节进行考察);湿法制粒变更为干法制粒(以颗粒得率、休止角和堆密度为指标);采用了动态超声逆流提取技术(以干膏收率、有效成分含量、指纹图谱为考察指标)。
②药材资源研究:美国要求药材来源稳定,产地有明确的GPS 坐标范围,III期临床试验和未来生产所用药材均应来自于所划定的GPS 坐标范围内。基地的选择以气候相似性为原则,道地产区相对广阔的一片区域,区域内含3 个或3 个以上药材基地,并遵循GACP 的规定,提倡使用家种药材,保证药材的基原单一。连花清瘟胶囊组方中的所有药材均已固定产地区域和核心基地,并采用传统的植物学鉴定和现代的DNA 条形码技术相结合保证基原准确,在基地管理过程中遵循GACP,由专人收集种植、管理、采收加工,提供了基础数据和影音资料。
③CMC 研究:USFDA 建议在现有条件下,尽量采用多种分析和技术手段,保证制剂间成分、纯度、质量、效力等一致,避免药物疗效及安全性受影响。USFDA 也强调严格的质量控制应从植物原材料到植物药成品,从非临床研究到临床试验,贯穿始终。从药材源头、中间体到成品建立可溯源质量控制体系。采用液相、气相、红外光谱等对药材、中间体和成品进行指纹图谱和多成分含量测定研究。不要求列出所有成份,只要求对植物原料药质量平衡、有影响的大类成份进行研究,并在III期临床前提供资料[61]。
4 美国已批准植物药的药学研究情况
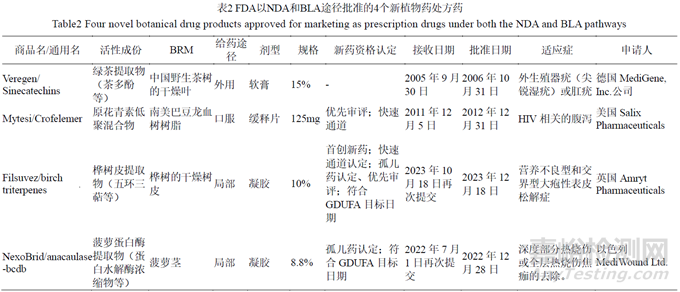
根据USFDA 公开的信息,截至2014 年,有超过600 个植物药申报pre-INDs/INDs,大约1/3 是商业申请,2/3 是研究目的;其中2/3 是单个植物原材料,1/3 是多植物原材料,大部分只开展了II期临床试验,只有少数进入了III期临床试验。其中申报的适应症主要有肿瘤(34%);麻醉、镇痛和上瘾(10%);皮肤病和牙科疾病(9%)等[62]。截至目前,USFDA 批准的新植物药处方药有4 个[63],见表2。4 个产品中3 个是以NDA 途径获批,1 个是以BLA 途径获批。给药途径和剂型以外用或局部皮肤用凝胶或软膏为主,只有一个是口服缓释片剂。除了Veregen 之外,其余3 个产品均获得了多项新药资格认定。
Filsuvez 和NexoBrid 在首轮审评时均未获得批准。Filsuvez 首次申请是在2021 年3 月30 日,2022 年2 月26 日收到完全回应函(complete response letter,CRL),其中指出该申请未能提供支持Filsuvez 治疗可以加速伤口愈合或减轻总伤口负担的充分数据。为了解决这个问题,需要提交有关该产品治疗EB(大疱性表皮松解症)或EB 人群特定亚集的额外的确证性有效性证据;Nexobrid 于2020 年6 月29 日首次提交申请,由于产品质量(product quality,PQ)和科学调查办公室(OSI)方面的诸多缺陷于2021 年6 月25 日收到CRL。从获益-风险评估的角度,关于PQ 缺陷,药物质量办公室(office of product quality,OPQ)的结论是,提交的数据不足以支持本品的生产是良好控制的,生产的最终产品在货架期内是纯净的、有效的。OPQ 建议向申办方签发CRL 概述其缺陷并提出支持本品批准所需要的信息和数据。其缺陷包括BRM 的真伪鉴定、菠萝蛋白酶的特殊生产工艺和原料药微生物控制、制剂微生物控制、制剂质量方面的化学、生产和控制等问题。此外,批准前需要对原料药中间体生产车间、原料、制剂以及凝胶溶媒生产车间进行检查,但由于旅行受限,USFDA 无法在这轮审评中开展检查。在本品再次提交后,OPQ 和OSI 方面的问题得到解决,审评结论均建议批准该申请,见表2。本品于2023 年10 月16 日提交增加儿童人群的申请,2024 年8 月15 日获批。
根据 USFDA 信息公开与保密的规定,NDA 不得公开的信息包括生产方法或工艺、质量控制程序等[64]。本文基于4 个已批准植物药产品的药学方面有限的公开信息进行总结,概括植物药的药学方面需要关注的主要问题,举例如下。①在BRM 方面,种属的鉴别(鉴别种、亚种和栽培品种等,区分易混品,提供真品证书)、采收地理位置(eco-geographic regions,EGRs)、加工过程的控制等;GACP符合性,对采收人员的培训,对BRM 建立质量控制方法,对农药残留、黄曲霉毒素、重金属等杂质进行控制;采取措施,加强环境的保护;提供数据说明BRM 的天然变异程度。②应建立原料药和制剂稳定性指示性的含量测定方法,并规定上下限,可接受比化学药更宽泛的含量限度,但需要提供充分的依据。③在质量控制方面,应建立杂质检查方法,并制定限度;建立生物测定法确保批间一致性;建立溶出度和溶出曲线检测方法。④对照品方面,需提供对照品的检验报告、MSDS 等,对于自制对照品要提供确认方案等。⑤含量测定、鉴别等方法均应经过验证。⑥制定可重复和可靠的原料药鉴别检验方法、结构鉴定和其他表征,采用多种方法联合测定表征。⑦药学研究与临床有效性和安全性的相关性,采用与临床终点相关的含量测定方法和指标。
5 讨论与展望
根据对 USFDA 植物药相关指南、我国在USFDA 申报品种案例、USFDA 已批准产品的公开药学研究内容的分析,总结美国植物药注册药学研究的关键点和难点,具体内容如下:
5.1 申报过程中与USFDA 的沟通交流至关重要
各个研究阶段申请人可就相关的问题与相关审评部门召开咨询会或以其他途径进行咨询,例如:早期临床试验植物原材料在种植和采收可能的变化和/或工艺优化,III期临床试验处方的变化以及上市后种植基地、种植和采收规范或加工方法等方面变更是否需要开展桥接研究,以及需要开展的桥接研究或其他研究类型;早期临床试验向制剂中加入单体活性成分的相关事宜;植物药临床试验中使用的安慰剂与试验药物间有细微差别的相关问题;多个适应症是否需要开发不同的生物测定方法等。
5.2 分阶段准备药学研究资料
对于有人用经验的植物药制剂,在美国可直接开展II期或III期临床试验,II期临床试验一般无需提交详细的CMC 资料,但在III期临床试验启动前需要提交较完善的药学研究资料。故在III期临床试验前一般会开展工艺再提升、质量标准再完善、生产车间的升级改造、工艺验证等,以及开发生物测定研究及质量平衡法,以便变更的内容或建立的质控方法在III期临床试验得到验证。III期临床试验后上市申请前不建议再有重大变更(例如原料药的生产场地、药材和生产工艺等),以确保临床试验用原料药与拟上市用原料药的一致性,而无需开展桥接研究。上市后对药材产地、种植和采收规范、和/或加工方法上所做的变更,均需要开展桥接研究。
5.3 生物测定研究
生物测定评价贴近临床作用机制,通过将化学组成和活性建立关系,从而评价原料药的整体活性,它可作为生产过程质量控制评价、稳定性试验评价、不同阶段原料药相似性评价、上市后变更评价及批间一致性评价的重要指标。作为一种质控方法,需要开展方法学验证和系统适用性研究以确保方法重复性和可预测性。
5.4 确保制剂质量一致性
质量一致性评价包括对BRM 的控制、化学检测的质量控制和生产过程控制、生物测定和临床数据的综合评价,以证明上市样品与上市前临床试验用样品的疗效一致。BRM 控制包括实施DNA 条形码鉴定技术、固定产地和基原、符合GACP 等,GACP 无法覆盖的种植、采收、加工和贮存等过程应符合cGMP 要求;提取物和制剂生产控制,应符合cGMP 要求,建立生物测定法整体控制质量,通过多批次、多剂量关键性临床试验,确保研究药物批次间临床疗效一致。其中,GACP、cGMP 及临床试验都需要大量的人力物力和时间投入,是注册获批的难点。
5.5 植物药作为药品,具有常规药品的一般要求,同时也要遵循其自身特点
例如,在药品通性方面,资料提交要求与其他药物相同,需要以eCTD 格式递交注册申报资料,植物原料可以以DMF 的形式或作为IND、NDA 资料的一部分递交;采用稳定性指示性的分析方法作为质量控制方法;参照化学原料药方式设定提取物质量标准,提取物批次混合投料提高指标成分控制的精度;原料和制剂要符合cGMP 等。在遵循自身特点的方面,对植物来源起始物料的严格控制;在稳定性的显著差异方面可以接受更宽泛的显著性差异(>5%);接受含量均匀度采用装量或重量差异而非含量差异进行测定;制定生物效价方法测定提取物(原料药)整体活性;采用多剂量多批次临床试验考察不同药材批次、不同剂量的效应关系等。
我国中药药学方面的技术指南经过20 年的发展,在总体原则方面已与美国相关要求一致[59],虽然可能还存在着理念、生产体系等方面的差异,但相信在未来,若能选择未被满足的临床需求、疗效确切、处方简单、成份相对清楚、国内研究比较充分的中药在美国开展植物药处方药注册,将大大提高注册成功率。
参考文献
[1] FDA. What is botanical drug[EB/OL].(2025 -01-07)[2025-04-18]. https://www.fda.gov/about -fda/cder-offices-and-divisions/what-botanicaldrug.
[2] FDA .Frequently Asked Questions on Botanical Drug Product Development[EB/OL]. (2025 -01 questions-botanical-drug-productdevelopment.
[3] Freddie A H. Botanicals as “new” drugs: US development[J]. EPILEPSY Behav, 2015,52:338.
[4] FDA. Botanical drug development, guidance for industry[EB/OL].(2016 -12)[2025-02-01]. https://www.fda.gov/media/93113/download.
[5] 张万良, 胡泽萍,周立红,等.中国、美国和欧盟植物药注册法规和技术要求