您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-08-04 09:02
ICH S1B《药物致癌性试验》指导原则的主要目的是提供一种科学合理的方法来评估药物的潜在致癌性。传统上,欧盟、日本、美国、中国等监管机构要求药物致癌性评估通常需要在两种啮齿类动物进行致癌性试验,其中包括可能需要进行的2年大鼠致癌性研究。
2022年,ICH S1B指南增加了附录(ICH S1B(R1)),引入了“证据权重(Weight of Evidence, WoE)”方法。这种方法通过评估WoE因素来判断2年大鼠致癌性研究是否对人类致癌风险评估有价值。这一方案的提出,部分基于2013-2020年期间进行的前瞻性评估研究(PES)的结果。
更新指南的主要依据是回顾性分析了来自13家PhRMA公司的182种化合物数据集(盲法),以及76种国际癌症研究机构(IARC)1类和2A类化合物的数据集。研究发现,在对2年大鼠致癌性研究结果分析后,如果以下三种情况均不存在:1)慢性毒性研究中大鼠肿瘤形成的组织病理学风险;2)激素干扰或内分泌药理学的证据(某些药物可能通过影响激素水平或激素受体的活性,进而影响细胞增殖和分化,从而增加致癌风险);3)遗传毒理学阳性结果,则82%的研究结果预测为致癌阴性。而剩余18%的化合物中,大鼠的致癌阳性结果被认为与人类的相关性存疑。因此,满足这些标准的化合物被认为不太可能是大鼠致癌物。基于这些标准,已经可以对人体致癌风险进行充分评估,而无需依赖2年大鼠研究的结果。
此外,对255种化合物的数据集(非盲法)进行回顾性分析显示,药物的药理活性与6个月治疗后大鼠的组织病理学发现以及随后的2年大鼠研究致癌性结果之间存在关联。说明,更全面的药物靶点药理学的理解有助于提高对2年大鼠致癌性结果的预测准确性。另一项对289种人用药数据分析表明,基于药理学和组织病理学预测大鼠非致癌物的成功率为92%,而预测大鼠致癌物的成功率为98%。
这些回顾性分析支持了ICH S1B(R1)专家工作组(EWG)在2013年“Proposed change to rodent carcinogenicity testing of pharmaceuticals- Regulatory Notice Document”文件中提出的假设,药物的靶点相关药理学和信号通路的知识,结合毒理学数据,足以表征其致癌潜力,因此也足以判断是否开展2年大鼠致癌性研究对评估人类致癌风险增加额外价值。然而,此前尚未开展前瞻性研究来验证WoE方法的预测2年大鼠研究的结果与人类致癌风险的能力。此外,也没有信息表明药品监管机构(DRAs)和行业是否能够基于WoE评估的结论达成一致。
因此,FDA(美国)、PMDA(日本)、EMA(欧洲)、HC(加拿大)、SMC(瑞士)组织开展了一项前瞻性评估研究,在没有获得2年大鼠致癌性研究结果的前提下,确定这种WoE方法的监管可行性(Bourcier et al.2024)。这项研究的主要目标包括:1)确定WoE方法是否足够稳健,能够预测2年大鼠致癌性研究的结果和价值;2)定义哪些因素有助于WoE评估,从而得出2年大鼠研究是否对人类致癌风险评估有价值的结论;3)评估DRAs之间以及DRAs与制药企业之间在预测方面的一致性。研究对45份致癌性评估文件(CAD,WoE评估报告)和22份完整大鼠2年致癌试验报告进行了分析,涵盖18种不同靶点,11种不同治疗领域或适应症。
下表罗列了药物致癌风险分级,后续根据具体分级展开讨论。

首先看下根据45份CAD文件进行致癌性风险分类,制药企业和DRAs对于1级致癌物的分类是一致的,对于2、3a和3b分级不完全一致。这点也容易理解,2类需要开展致癌试验,制药企业和监管机构所持的立场不同,前者倾向于合理减少,后者则更倾向于合理控制风险,故前少后多。
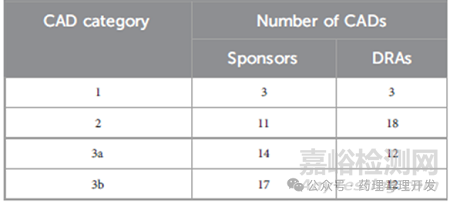
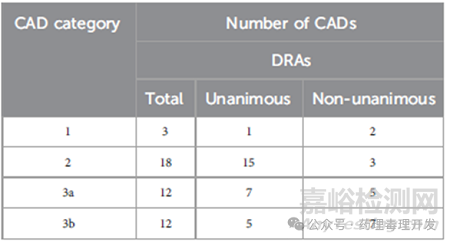
不仅药企和监管机构之间有分歧,FDA、EMA、PMDA、HC、SMC不同监管机构之间的理解也有差异。如下表所示,1、2、3a、3b分级中,监管机构达成一致的比例分别为1/3、15/18、7/12、5/12。提示,基于WoE路径进行药物致癌风险分级,有一定的主观因素在里面,制药企业、不同监管机构之间的意见和结论并不总是一致的。
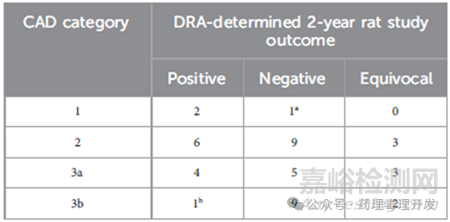
WoE毕竟是从靶点生物学、次要药效学、慢性毒性试验病理学、遗传毒性等角度进行理论分析,那么这种理论分析得出的结果与实际开展的试验结果一致性如何呢?如下表所示,阳性、阴性和结果不确定的数量分别是13、24和8例。以2级为例,监管机构按照CAD分级是18例,实际大鼠2年致癌试验结果显示,阳性6例、阴性9例、结果不确定占3例,所以这类化合物确实有很高的致癌风险,开展大鼠致癌性研究是有必要的。阴性率最高的是3b这个级别。
3b类
当预测的致癌风险对大鼠和人类均较低或不存在时,基于WoE评估的结果,将这类情况的化合物归类为3b类。在这种情况下,进行2年大鼠致癌性研究被认为不会增加额外价值。药企将17/45个案例归类为3b类,其中12个案例得到了监管机构的完全或部分同意。在这12个3b类案例中,11个案例的大鼠2年致癌试验结果为阴性,1个案例报告为阳性。不过,DRAs将两个药企指定为阴性的案例评估为不确定。特别讲一下3b中这个阳性案例,为什么会出现这个漏网之鱼呢?该案例中,药企和DRAs均认为肿瘤结果为阳性且与供试品相关,表现为子宫癌。对6个月的毒理学研究结果进行回顾性分析发现,在与2年致癌试验出现子宫肿瘤相近的剂量下,6个月毒理结果也显示子宫重量显著增加,且有异常内容物。不过,在进行CAD评估时,药企和DRAs均未将6个月研究中的子宫异常发现作为子宫肿瘤发生的风险因素,故归类为3b。这也提示我们,6个月慢性毒性研究中观察到的生殖器官重量增加(无论是否伴有组织学相关性)可能是长期给药后肿瘤发生的风险因素。总体看,被药企和DRAs同时归为3b类的化合物,致癌风险确实很低。
3a类
经WoE分析,认为化合物高度可能在大鼠中致癌,但通过已知且被广泛认可的机制证明在人体中不致癌,归为3a类。药企将14个案例划为3a这一档,DRAs则对其中12个案例完全或部分同意。在DRAs认定为3a类别的12个案例中,4个案例在2年大鼠致癌试验中显示为阳性,5例阴性,3例不确定。当然,这3例药企给出的结论是阴性,DRAs则认定为不确定。然而,所有阳性研究中观察到的肿瘤类型均不认为对人体具有致癌风险,原因在于这些肿瘤的发生机制被认为与人类无关,或者肿瘤是在高暴露量情况下出现的,故认为与人体不具相关性。
2类
当WoE评估表明人体致癌风险不确定时,这类化合物归为致癌2类。对于这种情况,2年大鼠致癌研究是有价值的。药企将11个案例归为2类,其中8例得到了DRAs一致同意。2类化合物的致癌不确定性主要来自其药理机制或化合物特异性毒理学发现,以及缺乏同类产品在大鼠致癌性研究中的信息。例如,有的化合物仅提交了3个月大鼠重复给药毒性试验,未提交6个月更长给药周期的数据。8例药企和DRAs意见一致的案例,实际的2年大鼠致癌试验结果是3例阳性,4例阴性,1例不确定。
另外,还有7例被药企建议归为3a或3b的案例,被DRAs归为2类。双方争议包括6个月大鼠毒理研究中获得的组织病理学检查与潜在人体致癌风险的相关性信息是否充分?对药物靶点的认识是否充分?举个例子,一款治疗炎症的丝氨酸/苏氨酸蛋白激酶抑制剂,药企认为应该作为3b类,原因包括:1)本品属于免疫调节剂;2)相同作用机制的同类化合物,致癌结果阴性;3)6个月大鼠和9个月猴重复给药毒理研究病理结果未见异常;4)遗传毒性试验结果阴性;5)雌性大鼠生殖组织的退行性变化被认为与人体无关。DRAs则不认同,观点如下:1)本品的免疫调节特点表征不充分,无法提示人体致癌风险;2)同类化合物观察到的毒性表现比较多样,无法外推到本品;3)需要进一步的免疫毒性研究数据支持;4)雌性大鼠生殖组织的病理学检查不充分。故,DRAs建议该化合物归为2类。
这7例被DRAs归为2类的化合物中,大鼠2年致癌性研究结果显示4例阴性,3例阳性或结果不确定。
1类
当WoE评估认为人体致癌风险非常高时,会指定为分类1。这种情况下,待测化合物的致癌风险比较确定,故2年大鼠、小鼠或转基因小鼠致癌性研究不会提供额外价值。药企提供的经WoE分析视为1类的化合物共3例,其中1例,所有DRAs均同意归为1类。另外2例,DRAs之间未达成一致,部分DRAs认为证据权重信息不足,仍然需要开展2年大鼠致癌试验。实际情况是,这1例多方达成一致的大鼠2年致癌结果阳性。另外2例DRAs有争议的案例,1例阳性,1例阴性。
最后
大部分3b分类化合物,WoE理论分析结果与大鼠2年致癌试验结果一致,致癌风险低。3a类化合物,按照WoE分析应为大鼠阳性,但人体不相关,实际大鼠致癌结果是有些阳性,有些阴性,阳性的与人体确实不相关。1类的则视为有比较高的致癌风险,实际结果是2例阳性,1例阴性。所以,对于1类、3a类和3b类,理论上不需要开展2年大鼠致癌结果,实际的试验验证要么与预期的阴性结果一致,要么与预期的阳性结果一致,要么预期阳性实测阴性,不开展大鼠2年致癌试验,能够大致覆盖风险。但2类的则不同,这类化合物需要开展大鼠2年致癌,实际结果是超过半数是阴性,即开展了本可避免的大鼠2年致癌试验。关于2类化合物归属这点,DRAs明显比药企更为保守,也可以理解,毕竟双方角度不同,前者更重视风险,后者还要平衡成本。最后,WoE是一项多角度多因素的综合评估,有些证据比较清晰、充分,多方容易达成一致,有些证据则不够清楚,主观因素掺杂其中,就会产生分歧。这些分歧不仅存在于药企和监管之间,不同国家监管机构之间的认识也并非完全一致。

来源:曾子像/药理毒理开发


