2025年5月5日,美国总统特朗普签署了一项名为“促进国内关键药品生产的监管减负”的行政令。该行政令的核心目标是通过简化监管流程,消除阻碍美国国内药品生产的监管障碍,推动药品制造回流美国,以增强美国在药品供应上的自主性和安全性。然而,这一行政令的“外紧”政策,即加强对外国药品制造设施的监管,给国内药品出口企业带来了新的挑战。根据行政令的要求,在未来90天内,FDA局长将制定并推进一系列改进措施,确保对供应美国药品的海外制造设施进行常规审查。此外,为了支持更多的检查活动,行政令还计划通过增加对外国制造设施的收费来筹集资金,这意味着海外药厂需要缴纳更多的费用来支持这些检查。
5月6日,FDA宣布扩大对外国生产设施的不事先通知检查(飞检)的使用范围。这一变化旨在确保外国企业接受与美国国内企业相同的监管标准,消除长期以来存在的双重标准问题。FDA局长强调:“长期以来,外国企业在检查前获得提前通知,而美国本土制造商则面临着严格的无预警检查。这一局面从今天起将被改变。”FDA将评估其政策和做法,以改进外国检查项目,确保自身成为监管监督的黄金标准。这包括明确FDA调查人员拒绝受监管行业提供的旅行住宿(包括住宿和交通安排)的政策,以维护监督过程的公正性。
FDA的这一举措意味着国内药品企业出口美国市场的成本和难度都将大幅增加。一方面,场地费用预计会显著上涨,直接加重企业的合规成本负担;另一方面,不事先通知的检查将使企业难以提前准备,要求企业必须始终保持高标准的合规状态。此外,任何记录造假或违规行为都可能被曝光,进而导致产品被拒、罚款甚至被禁止进入美国市场。例如作为出口美国的代表浙江华海药业在1月份接受的飞行检查,未得到事先通知,是FDA检查员到企业门口才知道的,所以此次检查最终检查结果整整11页的7条观察项,毫无疑问是OAI,后面也可以关注一下其整改及FDA的行动。
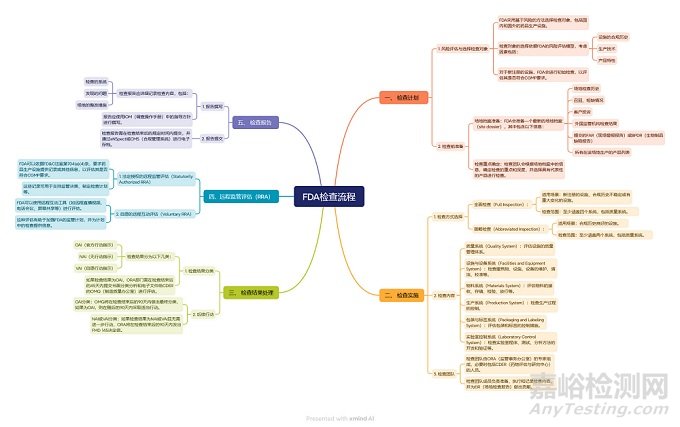
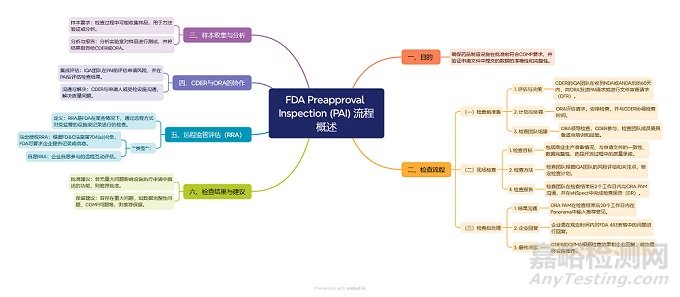
为了帮助国内药企更好地应对FDA检查,笔者特意整理了FDA常规检查(图1)和批准前检查(图2)的流程,希望能为大家提供一些参考。
▲图1-FDA常规检查流程
▲图2-FDA批准前检查流程
上述流程是基于FDA现行的符合性指南制作的, 检查由ORA(Office of Regulatory Affairs,法规事务办公室)负责,如为VAI/NAI,则由ORA发布最终决定信,如为OAI,则会进入CDER的OC (Office of Compliance,合规办公室),由其发布最终检查决定(该决定也可能会降级);对于PAI检查,则是由CDER的OPQ(Office of Pharmaceutical Quality)做出决定。
然而,需要注意的是,FDA的组织架构已经发生了调整。自2024年10月1日起,ORA被重组为OII(Office of Inspections and Investigations,检查与调查办公室)。重组后的OII继续负责检查工作,但大部分合规职能和人员已调整到各个中心,以简化操作并加快最终决定的做出(重组前要求在90天内做出决定)。
鉴于此,上述检查流程在未来可能会有所调整。笔者也已与FDA沟通交流确认,流程和时限没有变化,CDER合规部基于可用信息对OII分类为VAI和OAI检查做出评估及最终决定,而且最终检查决定的时间没有变,仍然是90天;至于何时会更新相关的指南文件,时间还不确定。笔者将持续关注FDA根据此次改组修订相应法规政策文件的后续进展,并及时为大家带来最新的信息。
FDA的检查流程与组织架构正处于持续优化与调整之中,其核心目标是进一步保障药品质量与公众健康。这一变革虽给国内药企带来了诸多挑战,却也为其提升合规水平与质量管理体系提供了契机。国内药企应积极主动地关注这些变化,及时调整合规策略,以从容应对可能出现的新挑战。药企需通过深入了解FDA的检查要求,建立完善的质量管理体系,加强员工培训,确保记录的真实性和完整性等措施,来提升自身的国际竞争力。只有这样,企业才能在满足美国市场严格监管要求的同时,赢得更广阔的发展空间。
此外,药企还应密切关注FDA对其他企业的检查结果,包括FDA 483、警告信、进口禁令等。需注意的是,FDA 483通常只有在收到3份或更多《信息自由法案》(FOIA)请求时才会发布,且发布时会删去商业机密或保密商业信息。FDA也可能在特定条件下主动发布这些文件。
相关检查数据与记录可通过以下网址查询:
● FDA Data Dashboard for Inspections
● OII FOIA Electronic Reading Room
通过分析这些信息,企业能够更好地把握FDA的检查要求与趋势,从而为自身的合规发展提供有力支持。