您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-01-25 22:43
带磁性的核在外加磁场中会产生能级裂分,吸收对应波长的电磁波就会发生能级的跃迁,停止电磁波照射后通过自旋晶格弛豫回到平衡态,产生核磁共振效应。经过核磁共振科学家多年不懈的努力后,二维核磁技术终于在20世纪70年代开始走向成熟,走向应用,典型的有同核COSY、DQF-COSY、TOESY、NOESY、ROESY技术,异核HMQC、HMSC、HSQC技术,另外还有研究核与核之间相互作用的NOE技术[1]。新药发现的一个主要途径是化合物文库和天然化合物的的筛选以及为了活性性质优化的大量结构类似化合物的合成。这些过程耗费大量人力物力,而且风险还很高,而这些技术为研究蛋白质的结构和小分子与蛋白质之间的相互作用带来了极大的便利。从20世纪末核磁共振技术首次成功运用于先导化合物的发现过程开始,一场新的药物发现技术革命悄然诞生。以下我们主要介绍核磁共振技术在先导化合物筛选过程中的开创性应用实例。
1、FK506结合蛋白抑制剂先导化合物的发现[2]
FK506结合蛋白和FK506结合,抑制钙调磷酸酶和阻止T细胞活化,为了寻找FK506和结合蛋白结合的抑制剂,Fesik等人避开了传统的筛选路径,开发了一套崭新的药物先导化合物筛选发现途径,具体总结过程如下图所示:
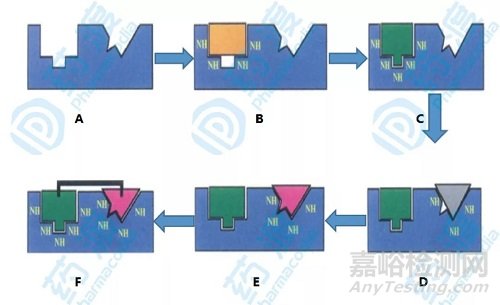
图1.基于核磁共振技术的分子设计步骤示意图
上图A是靶标蛋白的三维结构,通过第一轮小分子化合物配体筛选得到B,小分子与蛋白的结合作用可以通过N同位素标记的HSQC技术观察酰胺质子和氮原子的化学位移来识别,由于蛋白是N同位素标记,而化合物没有进行N同位素标记所以小分子底物不会干扰蛋白化学位移信号,从而提高了信噪比。第一个配体得到后经过一系列的药物化学修饰得到优化后的C,下面的目标则是寻找结合蛋白相邻位点的配体,确定的依据仍是核磁谱图中化学位移变化的相关情况,第二个配体粗选后继续经过药物化学的修饰得到活性最高的小分子化合物如上图E。在两个先导配体被选定后,通过核磁共振或者X-射线衍射技术得到三元复合物在空间的相对位置和小分子配体的空间取向,通过设计合适的连接子将两个配体片段连接起来得到最后的先导化合物如图上图F。
在FK506结合蛋白的抑制剂筛选过程中,第一轮筛选发现化合物文库发现如下图化合物1具有最高的活性,化合物1不经过进一轮的优化作为第一个配体,可以通过酰胺化学位移的变化断定化合物1的结合位置和FK506哌啶酸部分的结合位置一致。在化合物1饱和的条件下,继续寻找与相邻结合位点结合的小分子配体,通过对化合物库的筛选初步得到化合物2具有一定的活性,通过对在化合物1饱和的情况下化合物2加入后化学位移的变化,可以断定化合物2和化合物1的结合位点相近。
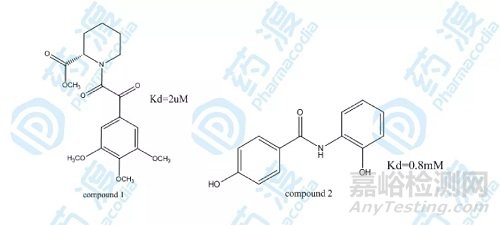
图2.化合物1和2的结构图
化合物2经过经过进一轮的构效关系研究得到最终高活性的化合物3。
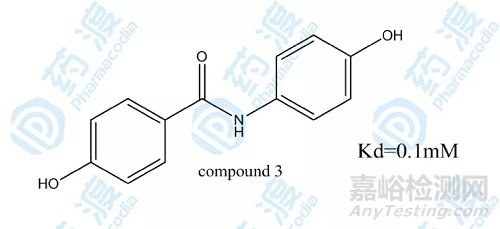
图3.化合物3的结构图
通过基于同位素滤波核磁共振试验建立三元复合物的空间模型,化合物1的甲酯结构和化合物3的苯环上的羟基结构在空间位置上比较接近,设计连接子跨过空间连接两个小分子配体,要保证设计的连接子不影响小分子配体在空间的位置和结构取向,设计如下图的分子结构。
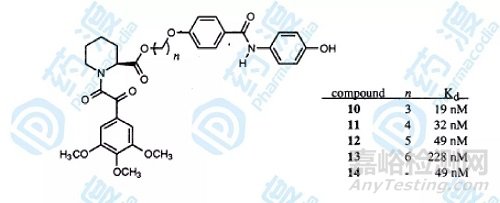
图4.连接子连接后的化合物结构和生物活性图
最终结果显示化合物10具有最高的活性,化合物14的分子式如下图,它采用了另一种连接方式,活性也达到了49nM。
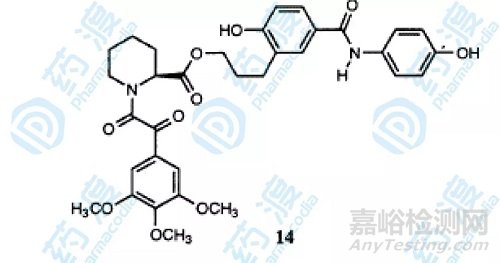
图5.化合物14结构图
为了观察两个小分子配体在连接之前和连接之后与结合蛋白的结合作用部位的变化,分别进行NOE研究,结果发现化合物1和化合物14的哌啶酸部分和三甲氧基苯基部分结合部位一样,与蛋白形成疏水相互作用,另外化合物3与化合物14的结合位置相似,化合物3的苯甲酰环与蛋白中Gln53,IIe56,Arg57的相互作用在化合物14与蛋白的相互作用中没看到,这又说明了尽管二者作用位点相似,但连接子的引入确实影响了化学位移的变化。
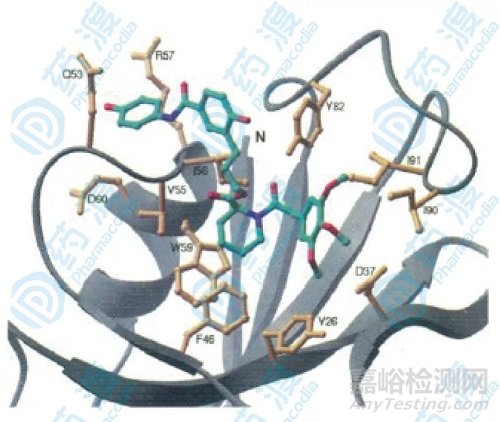
图6.相互作用示意图,黄色部分为结合蛋白和配体NOE效应的部分
从以上的实验可以看出化合物1的Kd=2uM,化合物3的Kd=0.1mM,当利用连接子巧妙地连接两个小分子配体后,得到的化合物活性都在nM级别。两个配体可通过不同的连结子(linker)连接起来,这种连接可以从焓变和熵变对结合能的贡献上得到理论支持[3],这使得先导化合物发现效率实现了一个质的飞越。
2、基质溶解素(MMP-3)非肽类抑制剂先导化合物的发现[4]
基质金属蛋白酶(MMPs)是锌依赖的家族内切蛋白酶,包括胶原酶,明胶酶和基质溶解素。该酶家族参与基质降解和组织重塑,并且当过表达或失调时与关节炎和肿瘤转移等病症相关。尽管已经发现了基于底物特异性设计的有效的基于肽的抑制剂,但是许多这些化合物表现出差的生物利用度。已发现几种非肽天然产物可抑制这类酶,如聚天冬氨酸和四环素衍生物。然而,这些化合物仅表现出中等的抑制活性。利用核磁共振技术S. W. Fesik等人成功的找到了对基质溶解素活性很高的先导化合物。
由于许多已知的MMP抑制剂含有异羟肟酸部分,用HSQC谱方法从化合物库中筛选到乙酰羟胺(CH3CONHOH)对基质溶酶的亲和力很弱,Kd=17mM,由于它的分子较小以及在缓冲液中的溶解度较大(大于500mM),未作任何优化选作第一配体。
在饱和量的乙酰羟胺存在下,用HSQC谱方法发现联苯及其类似物可以结合基质溶酶Kd在mM范围,经过近40余个联苯化合物的结构-活性研究比较(如下图),发现3'-氰甲基-4-羟基联苯(26)与基质溶酶的亲合力Kd达0.02mM。
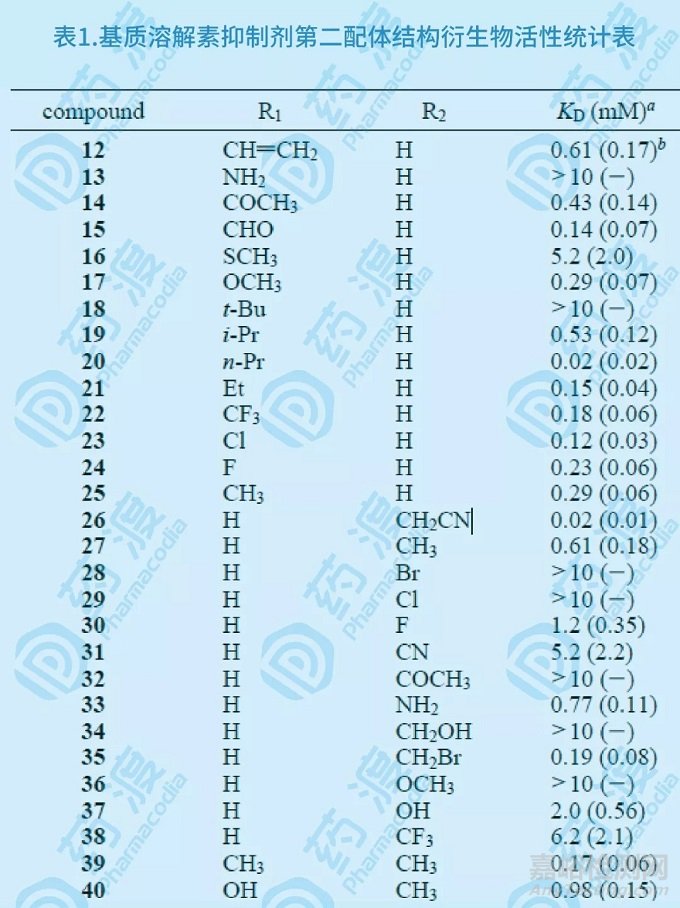
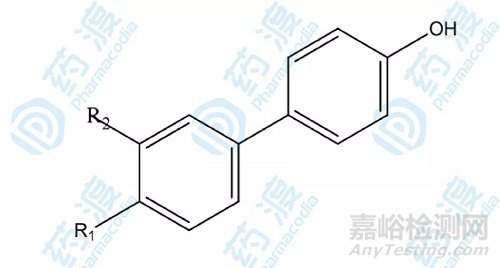
图7.基质溶解素抑制剂第二配体结构衍生物示意图
通过对乙酰羟胺,3'-氰甲基-4-羟基联苯类似物以及基质溶酶的三元配合物的NMR研究,设计合成了11个化合物(45~55),生物活性实验证明,与原有单体化合物相比,所有化合物的活性均有增强。对基质溶酶最具抑制活性的化合物50和53其IC50分别为25和15nM,活性增强近千倍。
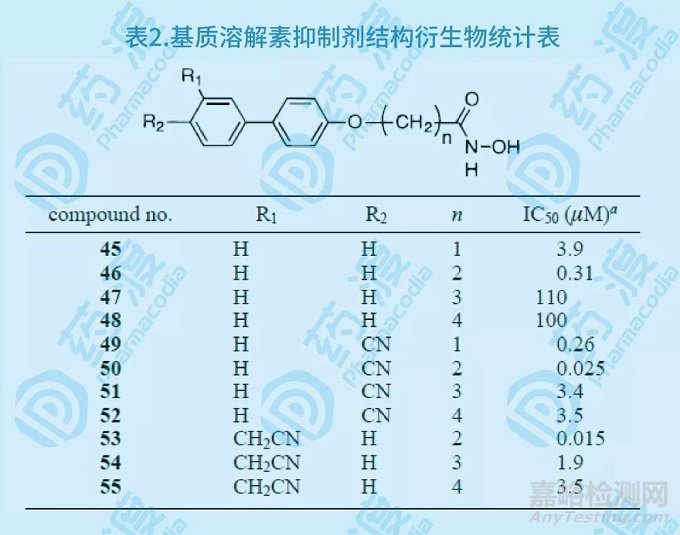
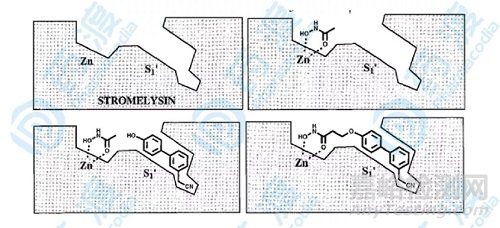
图8.基质溶解素抑制剂药物设计概括图
3、“SAR by NMR”和组合化学的对比
“SAR by NMR”方法在概念上类似于组合化学,然而这两种方法有本质的区别,在组合化学中发现配体需要制备测试大量的化合物,不幸的是,合成方案的选择和生物测定往往既困难又费时,进而相对统一的条件需要限制了有用的分子构建模块及化合物的多样性。在“SAR by NMR”方法中,配体已预选进行了优化,所以只需合成少数几个化合物即可。另一方面“SAR by NMR”方法不同于组合化学,筛选小分子化合物的范围仅仅受到水溶解度达mM浓度的限制,结果可以收集到多种多样的化合物样品[5]。
总结
本文介绍了核磁共振技术在新药研发先导化合物发现中的运用,需要注意的是以上这种核磁共振技术需要大量15N同位素标记的结合蛋白,另外结合蛋白的分子量大都在40KD以下等一系列技术限制,不过可喜的是目前已经开发出了两种不需要15N标记蛋白的新方法-一维驰豫编辑和扩散编辑NMR方法[6],弥补了这种方法的不足。
SPA(闪烁分析法)存在着放射性污染的环保问题,荧光分析法存在着底物浓度效应,荧光猝灭效应,化合物自身荧光干扰等问题,高通量筛选过程中会出现假阳性、假阴性,自动化程度高可是先导化合物发现效率低,而计算机辅助药物设计则存在着力场选择简单,计算过程繁琐,与现实情况符合性存疑等问题。新药研发的技术变革从未止步,相信随着核磁共振技术的进一步发展,必将继续提高先导化合物的发现效率,使得整个新药研发周期缩短。
参考文献:
[1] 毛希安.现代核磁共振实用技术及应用[M].北京:科学技术文献出版社,2000:251
[2] Discovering High-Affinity Ligands for Proteins:SAR by NMR[J]. Suzanne B. Shuker, Philip J.Hajduk, Robert P. Meadows,Stephen W. Fesik*. Science . 1996(274)
[3]Olejniczak E T, Hajduk P J, Marcotte P A, et al. J. Am. Chem. Soc., 1997, 119(25):5828.
[4] Hajduk PJ, Sheppard G, Nettesheim D G, et al. J. Am. Chem. Soc. 1997, 119(25):5818.
[5] 核磁共振技术在构效关系研究中的应用[J].化学通报,2001(1)
[6] Hajduk PJ, Olejniczak E T, Fesik S W. J. Am. Chem. Soc., 1997, 119(50):12257.

来源:药渡


