您当前的位置:检测资讯 > 实验管理
嘉峪检测网 2022-01-18 20:27
大家在做试验的时候,如果仔细查看色谱图,就会发现几乎每一个色谱峰都有一定程度的拖尾现象。有的比较严重,对有关物质检查会造成一定的影响。
这本不是问题,但是当拖尾严重到一定程度,就会影响我们的分析结果,所以今天就让我们来谈一谈关于液相色谱峰的拖尾问题。同时对气相色谱峰拖尾产生的原因及解决方法,进行简单的阐述。
什么是拖尾峰?
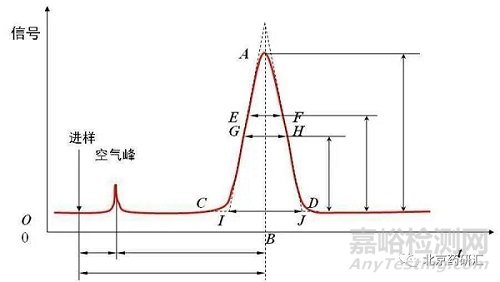
前沿陡峭,后沿较前沿平缓的不对称峰,称为拖尾峰。下图就是一个拖尾峰,我们会发现这个峰的右半边峰宽是大于左半边峰宽的,尤其在基线处更加明显。
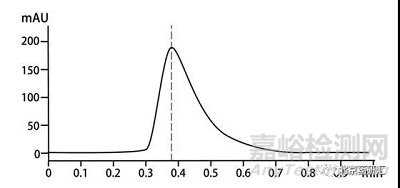
那如何评价色谱峰的拖尾呢?绝大多数情况下制药行业的从业人员会选择用美国药典(USP)中拖尾因子(Tf)来评价而其余的分析行业内人员则会选择用对称因子(As)来评价,拖尾因子和对称因子到底有什么关系?
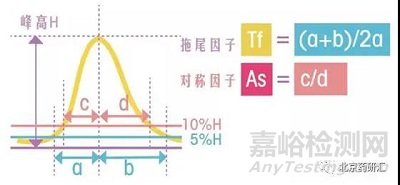
其中,拖尾因子Tf=(a+b)/2a,其中a和b分别是5%峰高处的左右半峰宽
对称因子As=c/d,其中c和d分别是10%峰高处的左右半峰宽
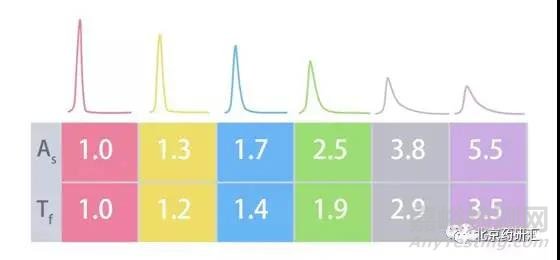
如果a=b,c=d,那么拖尾因子和对称因子的数值都等于1,也就是该峰不拖尾。实际工作中,当拖尾因子和对称因子小于2时,这两个值是非常接近的,如下图所示。
拖尾峰会对工作造成什么影响?
1. 影响峰高的计算
先来看看第一个峰(Tf=1.0)和最后一个峰(Tf=3.5),它们的峰面积虽然是相同的,但是由于最后一个峰拖尾很严重,所以它们的峰高相差三倍。当我们使用峰高来计算最低检出限时拖尾峰的负面影响就会比较明显。
2. 影响峰面积的计算
当我们对色谱峰进行积分来计算峰面积时,峰的起点和终点通常情况下通过色谱峰的斜率来确定。色谱峰越拖尾,它的尾部基线越平缓,终点就越难确认。这一点在基线波动较大时体现的更加突出,那么它就会影响我们积分得到的峰面积。
3. 影响对某些痕量组分的确认
在药典计算有关物质的相关要求中会提到,峰面积达到主峰面积0.1%的组分都要参与计算。
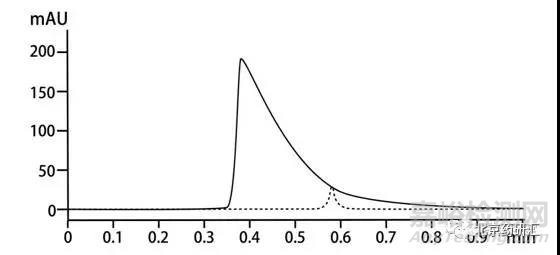
那么当一些小峰和前面的峰分离度不是很好时就会容易被隐藏在前面峰的拖尾处从而无法参与计算。
综上所述,拖尾峰会对色谱分析的定性定量结果都会产生影响。
产生拖尾峰的原因是什么?
1、拖尾峰的形成是因为在色谱分离时出现了一种以上的保留机制,并且其中一种保留机制是超负荷的。在日常分析中大多数情况下会使用反相色谱。以常用的C18柱为例,C18是被键合在载体硅胶表面的,而硅胶是由硅和氧聚合而成的,所以它的表面会出现各种形态的硅羟基。
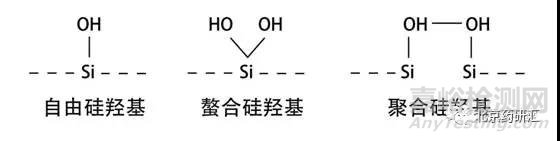
虽然理想的状态是流动相中的样品只和固定相C18发生作用。而实际上一半以上的硅胶表面并没有被键合上C18,所以硅胶表面会裸露出大量的硅羟基(Si-OH)。
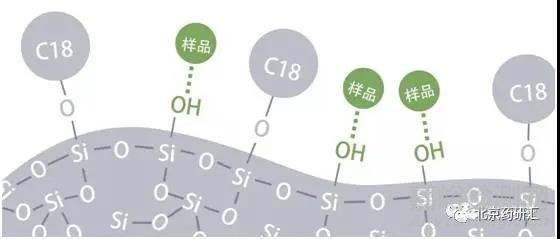
而单独的自由硅羟基会和流动相中的样品发生作用,形成拖尾。于是在最近的十几年中,色谱柱供应商致力于生产出高纯度的硅胶载体。由于新型硅胶在无金属离子环境下制造,所以表面不会残留金属离子也会减少拖尾。另外,硅胶表面尽可能减少自由的硅羟基,未与固定相键合的硅羟基尽可能形成螯合或聚合状态,用来减少拖尾峰的产生。
2、当色谱柱经历过高温、强酸,或者一些极端使用方法导致固定相流失较多后,色谱柱中的自由硅羟基也会变得越来越多从而更加容易造成拖尾峰。
3、拖尾峰的形成也有可能是因为柱外的死体积较多,导致样品在死体积处发生了滞留和扩散。
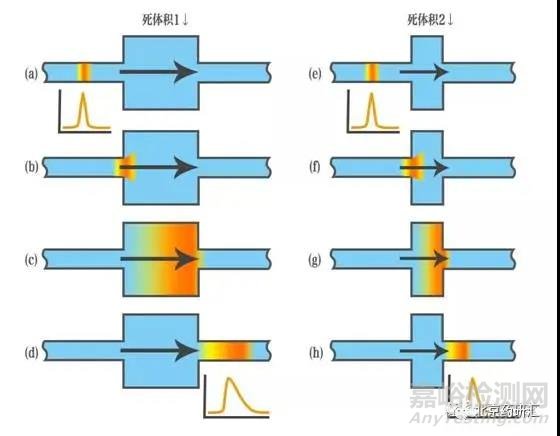
4、当样品中出现了碱性化合物,碱性基团更容易与色谱柱中的硅羟基发生作用形成拖尾峰。
注:气相色谱造成色谱峰拖尾的原因
前沿陡峭、后沿较前沿平缓的不对称峰,称为拖尾峰。气相色谱中,常见的吸附色谱法(利用吸附剂表面对不同组分物理吸附性能的差别,而使之分离的色谱法称为吸附色谱法),如果吸附等温线为非线性,当进样试样量超过一定数量时就会出现拖尾峰;分配色谱法(利用固定液对不同组分分配性能的差别,而使之分离的色谱法称为分配色谱法),如果载体表面具有活性作用点,试样量超过柱负荷或进样方法不当等,都会出现拖尾峰现象。什么原因会导致色谱峰拖尾?
引起色谱峰拖尾的原因比较复杂,如柱子两端安装不正确,没有达到进样口分流点和检测器处尾吹点位置;或安装好后又在接头处断裂;柱外死体积较大;尾吹气流量小,样品在柱内或系统内壁非线性吸附;汽化室污染等原因都容易造成拖尾。具体原因如下:
①进样量过大;
②进样器污染或汽化室中的衬管堵塞;
③衬管未脱活,造成待测物被吸附后逐步释放;
④载气流速过高;
⑤载气系统漏气;
⑥色谱柱安装不正确;
⑦色谱柱严重流失或污染;
⑧柱温太低或高于溶剂沸点温度;
⑨汽化室死体积太大;
⑩进样口汽化室温度太低。
如何改善拖尾峰?
1 与化学有关的拖尾问题 ?
(1)流动相中,加入30ml的三乙胺(用于碱性化合物)或醋酸胺(用于酸性化合物), 未知化合物加醋酸三乙胺;
(2)如仍然拖尾,将三乙胺换为二甲基辛胺(或醋酸二甲基辛胺);
(3)降低进样量至1μg。
2 与色谱柱有关的拖尾问题
(1)如柱头处有强保留的样品组分积聚,反相柱可用20倍柱体积的96%的二氯甲烷与4%甲醇,加1%氢氧化铵混合液冲洗;正向柱可用甲醇冲洗;
(2)使用保护柱。
3 与HPLC系统有关的峰拖尾和峰加宽
(1)进样体积过大,(通常≤25μL);
(2)进样阀与色谱柱及检测器之间的管路体积过大
(3)检测器流通池的体积过大。
当然气相又有点区别
还有要是其他峰不拖尾只有主峰拖尾那就可能是过载或与物质本身性质有关
注:如何改善气相色谱造成色谱峰拖尾
考虑到引起色谱峰拖尾的原因较复杂,可从以下几方面来分析解决。
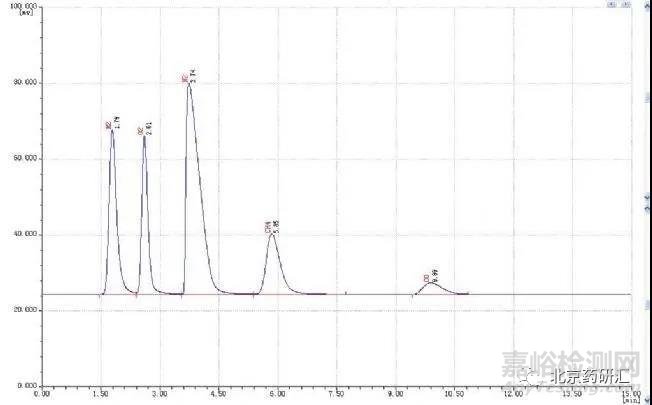
①进样量检查是否太大,减少进样量,观察色谱峰拖尾改善情况。
②进样口检查进样针针尖碰到并破坏了衬管内的填充物,堵在柱头,也会导致色谱峰拖尾,应从衬管中取出部分填充物、清理掉破碎物,或使用无填充的衬管。
③衬管脱活进样口衬管上的活性位点可能吸附待测组分导致出现拖尾峰,并可能损失灵敏度和重现性。对于不分流进样或分析极性很弱的化合物时,使用脱活衬管能尽量避免拖尾。当峰拖尾情况发生时,应及时更换衬管,尤其对于痕量分析,全新的衬管比清洗后并重新脱活的衬管在避免色谱峰拖尾上表现更有效。
④色谱柱温度超出色谱柱承受上限的温度会造成固定相和膜表面的加速损坏,这样会造成色谱柱的过分流失、降低柱效(分离度),尤其在有泄漏或载气中氧含量较高时,过度加热会大大加速并永久地损坏色谱柱,这样待分析活性组分的色谱峰就容易形成拖尾。
⑤色谱柱污染应切去色谱柱前端被污染的0.5~1m,必要时还需更换进样口衬管、隔垫,清洗进样口,严重时需要更换色谱柱。
⑥色谱柱位置在进样口中的位置不正确,泄漏或柱端切割不平整,均会导致色谱峰前伸或拖尾,应用专用色谱柱切割刀将柱端切割得干净而平整,重新按照安装尺寸安装色谱柱。
⑦进样方式不分流进样或柱上进样时,溶剂效应显著,应降低初始色谱柱温度,这样可使保留值增加、峰拖尾会减弱。
(4)案例分析
①衬管受污染和浓度过大引起的拖尾正己烷为溶剂配制有机氯农药标准溶液,用GC-ECD检测,各化合物色谱峰都有拖尾,观察仪器EFC,参数正常,气体无泄漏;检查进样垫,无泄漏;检查衬管,衬管内壁有黑色污染物,更换成新衬管,同样的色谱条件下进样,色谱峰拖尾有所改善,响应也有所提高;检查进样量,与以往无差别;将溶液浓度从2.0μg/mL稀释为0.1μg/mL,进样,色谱峰拖尾消失,表明溶液浓度过大,亦易引起色谱峰拖尾。
②色谱柱污染引起的拖尾色谱柱使用较长时间后,因受污染、柱流失严重,柱效降低。如HP一5(30m×0.32mm×0.25μm)色谱柱,使用四年,进敌敌畏标准溶液(0.1μg/mL),峰拖尾情况严重,老化色谱柱与切割柱前端都无济于事。
换成DB一5(30m×0.32mm×0.50μm)色谱柱,也使用四年,已污染,进敌敌畏标准溶液(0.1μg/mL),亦有拖尾现象;换成无污染的色谱柱DB一1701(30m×0.25mm×0.25μm),同样的条件下,进敌敌畏标准溶液(0.1μg/mL),峰形尖锐、峰宽较窄,拖尾现象解决。
③基质标准溶液进样,改善目标峰拖尾农药噻螨酮分析一般采用HPLC方法。研究中我们发现采用GC—MS亦能分析,但是乙腈溶剂所配标准溶液进样,噻螨酮出峰严重拖尾且响应很低。
当分别采用QuChERs方法净化后的芒果空白和西葫芦空白提取液配制标准溶液进样时,能明显改善噻螨酮色谱峰形和响应,当然也看到噻螨酮的西葫芦基质标准色谱峰呈现一定的圆头峰,可能跟不同基质下,噻螨酮在进样口活性位点上的吸附解吸附速度有关。

来源:Internet


