您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-09-24 19:57
难溶性药物在做固体分散体的时候,某个批次在中试车间出现了外观不合格问题。原因调查后,决定后续返工还是重新生产时,出现了意见分歧。有人认为制剂产品无法进行返工;有人则认为中间产品外观不合格可以通过返工解决,并不影响产品质量。制剂产品到底能否进行返工呢?为了找到答案,作者查询了相关法规和指南,首先明确一下结论,制剂工序返工在法规上确实可行。
一、返工的界定
《药品生产质量管理规范(2010年修订)》第一百三十四条,制剂产品不得进行重新加工。不合格的制剂中间产品、待包装产品和成品一般不得进行返工。只有不影响产品质量、符合相应质量标准,且根据预定、批准的操作规程以及对相关风险充分评估后,才允许返工处理。返工应当有相应记录。
《药品生产质量管理规范(2010年修订)》第一百三十五条,对返工或重新加工或回收合并后生产的成品,质量管理部门应当考虑需要进行额外相关项目的检验和稳定性考察。
ICH Q7 的14.2对返工进行了介绍,通常可以将中间体或原料药重复既定生产工艺中的步骤进行返工,例如重结晶或其他物理、化学处理,如蒸馏、过滤、层析、粉碎。
2023年版药品GMP指南-质量管理体系“3.4.3返工、重新加工”对返工进行了定义,将某一生产工序生产的不符合质量标准的一批中间产品或待包装产品的一部分或全部返回到之前的工序,采用批准的原生产工艺进行再加工,以符合预定的质量标准。适用范围:制剂和原料药。制剂中常见案例:如将未混合均匀的中间产品返回原工艺进行再次混合。“3.5.4不合格品处理”提到,对于返工所得到的中间产品和成品,除按常规质量标准检验外,还需综合考虑不合格的指标及其偏离程度、工艺特点、药品特性等因素,对可能受到影响的质量属性开展额外检验,并开展稳定性考察,与常规产品开展质量对比研究,以全面评估返工产品的质量风险。对于有可能重复发生的返工,还需考虑以同步验证的方式开展工艺验证,以确认返工工艺的可靠性、重现性。
为了避免概念混淆,再看一下重新加工的定义,将某一生产工序生产的不符合质量标准的一批中间产品或待包装产品的一部分或全部,采用不同于原生产工艺进行再加工,以符合预定的质量标准。并且给出了重新加工的决策树,见下图。重新加工只适用于原料药,且必须进行工艺验证。
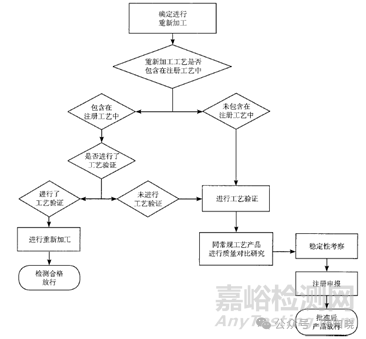
返工和重新加工的最主要区别是采用原工艺还是新工艺进行再加工。
二、返工的注意事项
参考2023年版药品GMP指南-质量管理体系文件,返工需要关注以下要求。
1、当导致物料或产品不合格的根本原因尚未调查清楚,无法据此对返工的风险进行有效评估时,不应进行返工。按偏差程序调查结束后,由生产部门按照原有工艺规程执行返工。
2、返工的产品必须指定唯一的可追踪的产品批号。
3、返工必须获得质量管理部门的预先批准,对返工进行的评估、额外检测及稳定性试验均应进行详细的记录。
4、商业生产产品进行返工的工艺需进行工艺验证,并经药品监管部门批准后方可实施。
由以上要求可知,制剂返工可行,但必须查明原因并得到质量管理部门的批准。如果是研发阶段的返工,能够查到根本原因,并做好预防措施,理论上可以按照偏差的方式管理。如果需要将返工写入工艺规程,希望解决商业化生产时偶尔出现的异常情况,则需要进行工艺验证。至于工艺验证需要做到什么程度,作者认为应该根据品种和工艺特点评估,结合案例情况在下一节进行分析。
三、返工的案例
对于返工来说,有可能发生在固体制剂生产过程中,液体制剂不太可能发生返工。
案例1、包装工序返工
根据返工的定义,我们见到最简单也最常见的返工是内包装。在瓶装或者泡罩包装时,可能因为某些原因导致一些内包装存在问题。有的时候,把内包装存在问题的样品作为废弃物;有的时候会取出药片或胶囊重新进行内包装。因为产品已经成型且内包装工序基本不影响产品质量,基于风险评估考虑,即便是返工进行内包装,也很少有公司对内包装返工进行工艺验证和/或稳定性考察。
案例2、混合工序返工
无论是预混还是总混,当发现混合不均匀时,一般情况可以通过延长混合时间达到混合均匀的目的。比如,总混加入润滑剂时,将润滑剂直接倒在颗粒表面后即刻总混,可能导致润滑剂第一圈转动时,部分润滑剂直接压实在混合机盖子内表面,进而导致没有完全混匀。这种异常情况的原因比较明确,但我们关心的是,能否继续混合以确保硬脂酸镁混合均匀。作者的建议是:主要看早期研究是否有混合时间对产品质量(压片硬度或溶出)影响的考察,能否覆盖掉再次混合的时间。如果有相关的考察数据支持混合时间延长,从风险评估的角度,可以返工继续混合,也可以不进行工艺验证。这也就是为什么中试批考察混合时间的时候最好做三个时间点,工艺验证的时候取中间时间点验证,工艺信息表中尝试申报时间范围,以便应对未来出现不可预料的偏差。
案例3、干法制粒工序返工
在做干法制粒工艺时,因工艺参数控制不合理或者设备因素导致产生过多细粉,细粉量超过一定范围将影响后续压片,此时可能会将部分物料进行二次干法制粒的返工操作。2023年GMP指南“口服固体制剂”对干法制粒这种细粉的操作,称为“颗粒回收”,干法制粒过程的颗粒回收操作应进行充分风险评估并建立相应操作规程,同时开展验证工作。这是在GMP指南中明确提到需要验证的工序,因为所有干法制粒工艺的产品,干法制粒工序基本属于关键工艺步骤。无论是参数控制不合理还是设备因素导致,基本上属于重复发生的事情,所以会在工艺规程中明确操作过程,并经过工艺验证和后期稳定性研究,干法制粒的返工也就有了合法的“身份”。
案例4、喷雾干燥工序返工
为了提高难溶性药物的溶出速率和生物利用度,通常会制备成固体分散体,现在越来越多的创新药采用固体分散体技术。喷雾干燥、流化床、热熔挤出是制备固体分散体的三种常用方法。
因为流化床制备固体分散体通常以辅料为底物,所以制备成固体分散体后,物料混合后变得复杂,已经无法进行返工,但是,无底物流化床制备固体分散体工艺除外(但是比较少见)。热熔挤出工艺的加热温度通常能够将原料药和载体粉末一同熔化,制备出来的固体分散体颗粒硬度较大,比较难返工到热熔挤出的螺杆内。喷雾干燥机制备固体分散体时,通常将原料药和载体粉末一同溶解在有机试剂中形成药液,药液雾化后瞬间干燥成固体分散体粉末。因此,后续仅讨论喷雾干燥工序返工。
喷雾干燥时,因为雾化喷枪堵塞或喷至内壁等其他原因,导致药液不能完全雾化干燥成粉末,可能会形成团块。此时,可能涉及到团块重新溶解再喷雾干燥的返工操作。团块未必没有形成固体分散体,有可能仅仅是外观不符合规定。但无法通过团块取样检测晶型规避风险,因为取样检测存在一定的概率,所以,最好的处理方式就是将团块物料返工。喷雾干燥工序的返工,有点类似原料药重结晶返工,均是将固体物料溶解后重新生产出符合质量标准的固体粉末。喷雾干燥返工可能会导致原料药两次受到高温处理,有关物质有增大风险(热熔挤出法经历的高温更剧烈,风险也更大)。因此,最好的策略是在小试研究阶段进行返工,然后考察稳定性,以支持中试期间可能出现的异常情况。如果需要支持工艺验证或商业化生产时的异常情况,则可以取部分物料在中试阶段或工艺验证阶段进行返工,考察0时的喷雾干燥粉末质量以及贮存期间的稳定性,不需要将喷雾干燥粉末制备成制剂后再考察产品稳定性。因为固体分散体是本工序步骤的直接产出物,能够反映出产品质量。
喷雾干燥通常采用有机试剂作为溶媒,所以需要控制溶剂残留,但喷雾干燥有时并不能将有机溶剂控制到合格限度,还需要进一步真空干燥去除溶剂。而残留溶剂的测量通常需要在QC实验室完成,时间较长,若溶剂残留不合格,可能会需要返工真空干燥。与案例2混合工序类似,建议研究阶段特意延长真空干燥时间,考察干燥时间延长对有关物的影响。有了研究数据支持,若真发生异常情况,需要返工,也便于质量部进行风险评估。
四、结语
对于偶尔可能发生的异常情况,调查原因后,基于对产品质量的影响和研究数据的支持进行风险评估,可以不进行工艺验证或部分验证,如混合工序、喷雾干燥工序。对于重复发生的情况,如干法制粒工序,则需要进行全套的工艺验证及稳定性考察。作者可能无法穷尽所有可能发生返工的情况,读者如有遇到其他情况,欢迎留言交流。
法规指南有的时候给了我们很明确的指示,我们应该按照现行版要求执行。当遇到法规指南没有明确指示时,作为药品研发人员和上市许可持有人,应该基于风险评估的角度决定做哪些研究,不能无条件求全,我们既要对产品质量负责,也需要考虑研发和生产的成本。

来源:药知晓


