药品标准是把握药品质量的标尺,药品标准中的检验项目需围绕药品的设计制定,检验项目中采用的方法为药典通用方法如干燥失重,重金属检查等无需对方法验证,但是特异性的检验项目的方法需有方法学验证内容以支持方法的可行性,如有关物质检查的HPLC方法,含量测定的HPLC方法等。方法学验证中容易出现的偏差,如验证结果未在Chp2020版四部通则的9101规定范围内,需要找寻闭环方法,使分析方法和操作方法更完善,顺利通过验证。本文结合验证实例对方法学验证中的偏差闭环给出一些方法,供同行参考。
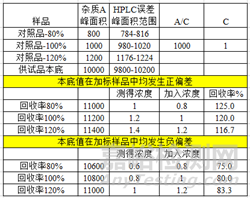
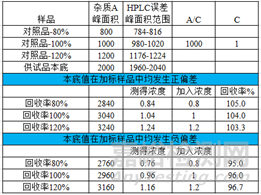
首先,偏差大咖是回收率未在要求范围内,举例:特定杂质A的限度要求为不得过3%,参考待测成分含量和回收率限度的规定,杂质A的回收率限度为92-105%,但是测定结果中80%水平的回收率结果分别为108%、90%、107%,未在限度要求的范围内;发生偏差后调查使用玻璃仪器等在校验范围内,操作无偏差,对照品使用得当,最后发现是供试品中的基质造成的回收率测定结果偏离限度要求。如果供试品本底响应值远大于对照品的响应值,在加标供试品溶液中仅仅是供试品本底响应值的偏差就会对回收率的结果造成较大影响,导致回收率结果偏离限度的范围,如下图所模拟的影响过程。倘若本底值和对照品值发生相同方向的偏差,测得的回收率结果将会出现更大程度的偏离。
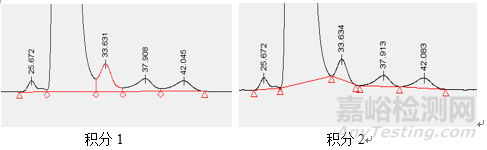
本例中的回收率偏高的原因是基质的影响,那么如果供试品的本底在定量限水平远远不会影响到回收率的结果,那么还有哪些原因会导致回收率偏高呢,排查仪器设备,玻璃器具,操作不当等在外,原因包括,①对照品影响,对照品开启时间较长吸潮,或者对照品的使用不当(部分对照品会标注干燥后使用),这样造成测得值的结果偏高,回收率偏高。②杂质B与主峰的分离度基线分离(分离度1.9),杂质B的限度为0.5%,回收率的限度要求为90-108%,如下图所示(杂质B的保留时间为33.6min),那么,积分方式的选择就对测定结果的影响较大,本例中以外标法定量,对照品溶液与供试品加标溶液的积分方式完全一致,模拟一下积分方式变化对结果的影响过程:杂质B的加入浓度为1,对照品溶液中杂质B的峰面积为5000,那么A/C为5000,供试品加标溶液中杂质B为6000,测得100%水平回收率为120%。当调整积分方式为峰谷到峰谷之后,对照品溶液中杂质B的峰面积为4800,那么A/C为4800,供试品加标溶液中杂质B为5000,测得100%水平回收率为104%。
③供试品溶液不稳定,供试品分析时长90min,在进样室中供试品不断降解成特定杂质C,导致杂质C的回收率异常高,可达200%左右。
由以上几个回收率偏差的例子得出闭环策略:a杂质的定量方法中,供试品本底值的响应水平需要远远小于对照品的响应值,可以通过供试品精制获得较小本底值的样品,通过供试品稀释减小本底值对测定结果的影响,或者改变定量方法为标准加入法测定;b对照品的使用中,需要提前了解对照品的理化性质,如引湿性等,避免因为对照品赋值不准造成的回收率结果偏差;c积分方法的选择,对照品和供试品溶液的积分参数如峰宽,斜率等直接影响峰面积的参数需要一致,在调整积分参数的时候优先以供试品为参考,同时兼顾主成分与杂质的积分合理性,避免导致杂质的积分过大,当然在积分方式影响较大的方法中,通过改变色谱柱。柱温或流速等方式优化方法更为重要;d供试品溶液稳定性的提前考察,必要时选择进样室控温的方式或者临用现制的方式进行方法验证;
其次:偏差大咖是含量测定结果偏高或偏低。举例:API的HPLC方法测定其含量,方法的专属性良好,含量结果的限度要求为98.0%-101.0%,但是测定的结果却总是偏低,测得值在96.0%。在偏差调查中发现仪器器具、试验操作等均正常,对照品的F值也在99.0-101.0范围内,仪器精密度良好,重复试验测试结果还是96.0%,对照品为刚开瓶,操作正确。后来考虑对照品的赋值存在问题,买到的API对照品为非法定对照品,在COA报告中赋值方式采取的是定量核磁的结果,经过其他品牌的对照品重新对供试品测定,发现含量的结果为99.0%,本次偏差是第一次采用的对照品的赋值偏低造成的测定结果偏低,可见非法定对照品作为含量测定的对照品风险是很高的,需要评估其赋值方式是否合理。举例:口服片剂的HPLC方法测定其含量,方法的专属性良好,含量结果的限度要求为95.0%-105.0%,但是测定的结果偏高,测得值在106.0%。本例调查结果是试生产时部分压片不准,造成平均偏重偏大。因为片剂的含量计算方式为含量(%)=((m测得值/m取样量)*平均片重)/标示量*100%,造成含量偏高的原因还可能是包衣在研磨过程中不易粉碎,出现取样不均匀,造成测得值偏高,进而含量偏高。
由以上的含量偏高的偏差得出闭环策略:a在原料的含量测定中,对照品的选择要慎重,对其对照品的赋值准确性需要进行评估后使用,必要时可通过精制原料自制工作对照品进行标定;b在制剂含量测定中出现异常偏高,需要考察工艺中的原因,还有含量用供试品的制备过程的优化,比如是否需要去除包衣等。
最后,偏差大咖是RSD不在限度要求的范围内,RSD是评价一组测定结果的精密度指标,之所以需要制定RSD的限度,是为了避免测定结果离散之后超出杂质的限度要求,影响测定结果的真实性,比如,限度为0.1%的杂质,在0时测定值为0.03%,平行6次,仅一次0.05%时,RSD为24.5%,在0时测定值为0.08%,平行6次,仅一次0.1%时,RSD为9.8%,虽然第二次的RSD更小,但是却有杂质超限的风险,因此在制定RSD可接受标准时,需要结合待测杂质水平,这也就是LOQ的RSD一般可以接受10%甚至15%的原因,那么LOQ的RSD依然超过限度造成偏差时,排查原因,操作正常,仪器器具正常使用,原因为杂质的灵敏度极高,S/N约为10时,峰面积仅900左右,并且相当于供试品的0.001%,因此较小峰面积很容易产生较大的RSD。另外一种容易出现RSD偏差的情况是重复性实验中,一般与RSD的可接受限度的制定相关。
由以上的RSD偏差得出闭环策略:a根据实测杂质水平,特别是一些单杂测定值很低,在评价重复性等RSD指标制定中可适当放宽,或者换绝对偏差,极差等方式评价;b定量限水平需要根据实际待测物质的灵敏度制定,不必固定于S/N在10附近。
综上列举了几种方法学验证中出现频率较高的偏差类型,并结合实例给出了闭环策略,希望可以给到同行工作中一些参考,同时也欢迎大家一起探讨。