您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-06-21 15:20
细胞周期失控是癌症的一个标志性特征,而CDKs在许多癌症中均过度活跃从而导致细胞增殖失控。随着CDK16研究的深入,包括证明CDK16表达的增加与肺癌和乳腺癌的总生存率降低有关以及相关抑制的筛选和发现,CDK16成为治疗癌症的潜力药靶,可谓是“柳暗花明”。但CDK16的抑制剂离治疗癌症的药物还有较大距离,可谓是“灯火阑珊。
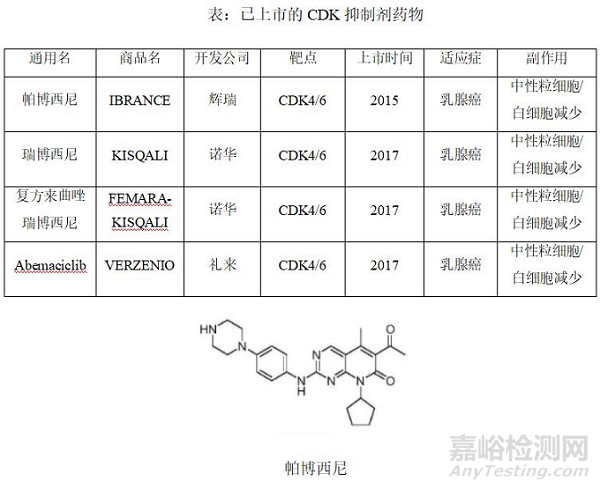
细胞周期蛋白依赖激酶(CDK)家族包括参与转录和细胞周期调节的多种激酶:现在发现共有21个成员,命名为CDK1-20(其中CDK11包括CDK11P85和CDK11P110)[1]。CDK抑制剂虽然已成功用于肿瘤临床治疗中,但其在基础研究中仍有很多未知,使该类抑制剂的疗效、适应证、敏感人群的选择等仍不确定。因此,目前全球仅批准了4款CDK抑制剂上市,均作用于CDK4/6靶点,主要以乳腺癌为适应症:帕博西尼[2](palbociclib,商品名:IBRANCE)是由辉瑞(Pfizer)公司研发;由诺华公司研发的瑞博西尼[3](商品名:KISQALI)和复方来曲唑瑞博西尼[4](商品名:FEMARA-KISQALI);由礼来公司研发的Abemaciclib[5](商品名:VERZENIO)。鉴于CDKs是癌症治疗中相当重要的靶点,控制细胞的周期,使癌细胞增殖受阻是癌症治疗中科学有效的策略;但另一方面现在CDKs中已有药物上市的药靶暂时只有CDK4/6,所以其他CDK的成员还有很大的潜力。随着CDK16的晶体三维结构被解析出来,与之相互作用的小分子抑制也投入筛选和研究,CDK16很可能成为一个令人瞩目的抗癌药物靶点。
CDK16的药靶潜力
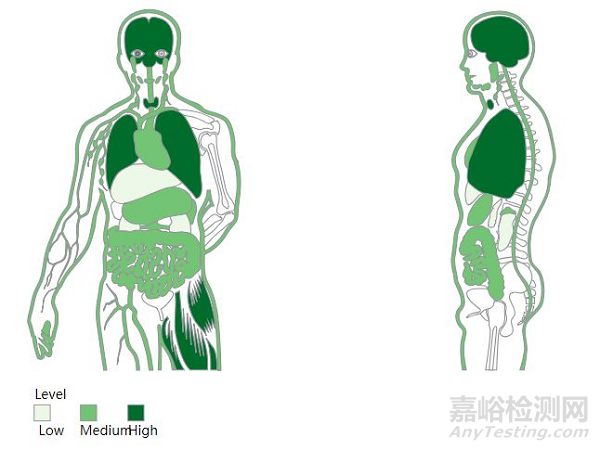
CDK16已被证明在各种细胞类型以及广泛的组织中广泛表达[6]。CDK16的最高表达水平在大脑和睾丸中,使用小鼠敲除模型的研究表明,CDK16在精子发生中是必不可少的,在轴突生长和细胞内小泡的调节中起作用。重要的是,由于其作为潜在药物靶点的相关性,最近的药理学的一些研究表明,CDK16表达的增加与肺癌和乳腺癌的总生存率降低有关[7]。CKD16的促肿瘤活性可能通过下调肿瘤抑制因子p27介导[8]。
CDK16在人体的表达情况[9]
根据它的表达情况以及它与癌症的相关情况,这个靶点是比较理想的药物靶点。首先它的表达的增加与肺癌和乳腺癌的总生存率降低有关[7],为药靶的成为提供了很大的可能性;另外CDK16不同于其他维持细胞生存基本功能的激酶,人体也许对于CDK16的要求是有限的,敲除CDK16基因的老鼠也不会出现死亡情况,也就是说完全缺失CDK16的情况老鼠还是能够存活,那么相关抑制药物作用于CDK16和可能不会影响到人正常的功能以及生命活动,但却对能增加癌症的存活率,这也是CDK16靶点的潜力所在;另外CDK16与细胞周期蛋白依赖激酶的成员一样对于细胞周期的进程非常重要,那么对于癌细胞这样增殖很快的细胞来说很可能是能有效抑制的,也就是说CDK16对于癌症的治疗是有有效性的。
CDK16抑制剂的研究现状
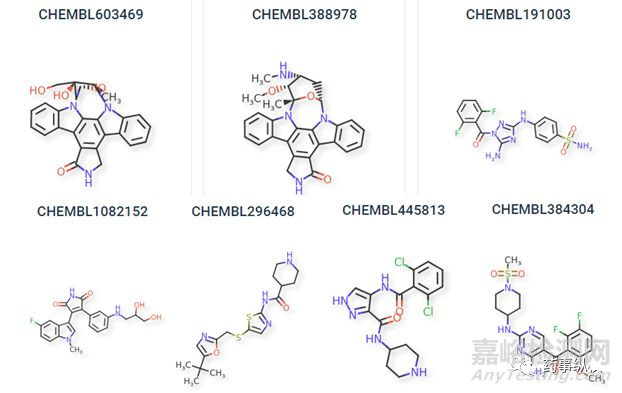
许多科学家都认为CDK16是一个具有潜力的抗癌靶点,进而开展了许多针对CDK16的药物筛选工作。7个小分子被报道为有可能的CDK16抑制剂[10][11],由于CDK16表达的增加与癌症的总生存率降低有关,这7种有效的抑制剂也许有治疗癌症的药物潜力。这些化合物的范围从非选择性激酶抑制剂R547(CDK16解离常数(Kd)值为0.5 nM)到CDK家族选择性抑制剂AT-7519和SNS-032(Kd值分别为1.1 nM和7.1 nM)。SNS-032最初作为一种CDK2抑制剂开发,通过激酶抑制筛选,SNS-032被证明对CDK16有抑制作用。从以下化合物有一些共性的结构存在说明也许要成为CDK16给抑制剂的结合造成了一些空间位阻。这几个CDK16的抑制剂主要是通过分子对接虚拟筛选出来的,接下来将可以进行药理的相关实验探索构效关系等,进而进行结构优化。并根据实际动物实验等数据可以设计出更加高效低毒的抑制剂。
7个有效的CDK16抑制剂
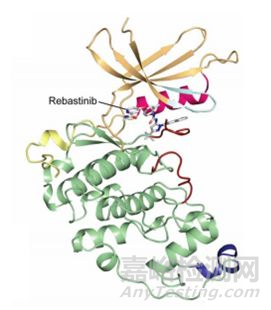
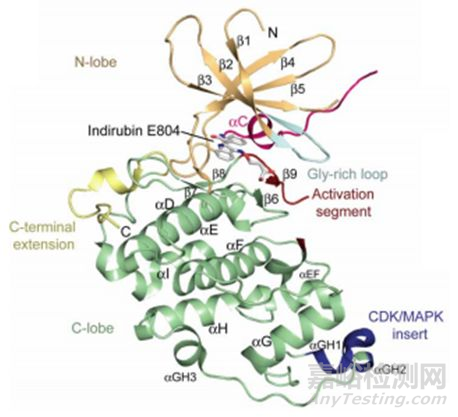
对于CDK16抑制剂的筛选工作,除了用分子对接虚拟筛选获得的以上的小分子抑制剂以外,还有用rebastinib(PDB:5G6V)和indirubin E804(PDB:3MTL)与CDK16共结晶获得了抑制剂和CDK16的蛋白复合物结构[12]。其共结晶的三维结构会展示抑制剂与CDK16的结合位点以及空间位置,在indirubin E804存在的共结晶中:indirubin E804大部分键是平面的,有三个氢结合到CDK16激酶的铰链区(kinase hinge region)。激活后的CDK16第305位的氨基酸:苯丙氨酸会翻转构象,进而在抑制剂周围形成笼状结构。因此,这种结合具有诱导的适应性,使激酶-抑制剂相互作用最大化。在 rebastinib存在的共结晶中:情况与indirubin E804的共结晶情况相似。这些晶体复合物结构提供了化学起点和关键的结构信息,复合物三维结构地深入分析有助于探讨药物-蛋白的结合和作用机制,有助于拓宽CDK16底物的范围,使设计选择性和有效的CDK16抑制剂成为可能。
CDK16和rebastinib的复合物晶体结构
CDK16和indirubin E804的复合物晶体结构
针对CDK16的小分子抑制剂的筛选工作证明了一些小分子对CDK16有较好的抑制作用;而一些抑制剂与CDK16共结晶的晶体结构为我们提供了更加精细的药物-CDK16相互作用的细节。这些研究对于抗癌药物的研发是非常重要的一步,也标志着CDK16抑制剂确实有成药的潜力,CDK16也成为了一个非常有潜力的药物靶点。
结语
细胞周期失控是癌症的一个标志性特征,而CDKs在许多癌症中均过度活跃从而导致细胞增殖失控。虽然目前全球仅批准了4款CDK抑制剂上市,均作用于CDK4/6靶点,主要以乳腺癌为适应症。但随着CDK16研究的深入,包括证明CDK16表达的增加与几种癌症的总生存率降低有关以及相关抑制的筛选和发现,CDK16成为了一个十分有潜力的药物靶点。CDK16的抑制剂研究和开发已经受到了重视,相关抑制剂的研究和开发也许在将来将会成为如同上市药物帕博西尼等一样为癌症的治疗带来希望。

来源:药事纵横


