您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-08-16 20:47
摘要
生物药物(biologics)近年来在全球范围内迅速发展, 其分析技术涵盖分子结构表征、纯度与杂质分析、功能及稳定性评价等多个方面, 是确保生物药物质量的关键。质量源于设计(quality by design, QbD)作为一种系统化方法, 在制药行业中被广泛应用于生产工艺的优化和确保最终产品的质量可靠性, 而基于QbD框架衍生发展的分析质量源于设计(analytical quality by design, AQbD)则为所开发的产品“分析方法”的质量赋予信心和提供保障。AQbD是以定义分析目标概况(analytical target profile, ATP)为起点, 通过风险评估识别和分析关键方法属性(critical method attributes, CMAs)及关键方法参数(critical method parameters, CMPs), 并利用实验设计(design of experiments, DoE)构建数学模型探索CMPs与CMAs之间的关系, 以建立方法参数的可操作设计区域(method operable design region, MODR)和分析方法控制策略。与基于传统理念开发的分析方法相比, 基于AQbD理念开发的分析方法在MODR范围内具有更高的稳健性, 能够减少因分析方法所导致的超趋势或不合格结果的产生, 从而实现监管灵活性并降低分析成本。本文综述了AQbD的工作流程及其在生物药物分析方法开发中的应用现状, 并探讨了该领域实施AQbD的机遇与挑战。
关键词
生物药物; 分析方法开发; 分析质量源于设计; 分析目标概况; 可操作设计区域; 分析方法控制策略
生物药物(biologics)是综合运用生物学、生物化学、免疫学及现代药学等原理与方法, 利用生物体、生物组织及其成分等生产的一类用于预防、诊断和治疗疾病的制品。常见的生物药物包括抗体药物、重组蛋白、疫苗、血液制品及细胞与基因治疗产品等。近年来, 生物药物以超越传统小分子药物的增速在全球迅猛发展。其分析方法涵盖结构表征、纯度与杂质分析、功能及稳定性评价等多个层面, 各类方法都独具特点和作用。通过综合运用这些技术, 可以全面评估和控制生物药物的质量与安全性。
“质量源于设计”(quality by design, QbD)是一种基于科学和风险的系统方法学, 最早由约瑟夫·朱兰于1990年提出, 强调产品质量问题与其设计密切相关。为推广QbD在制药领域的实施, 人用药品技术要求国际协调理事会(International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, ICH)与美国食品药品监督管理局(US Food and Drug Administration, FDA)及欧洲药品管理局(European Medicines Agency, EMA)联合发布了相关质量指南, 详细阐述了QbD的原则、工具及实际应用, 包括ICHQ8(R2)药品开发, ICHQ9(R1)质量风险管理, ICHQ10药品质量系统, ICHQ11药品原料的开发和制造, ICHQ12药品产品生命周期管理的技术和法规考虑, ICHQ13药品原料和制剂的连续制造。QbD运用设计工具与统计分析, 优化工艺并降低生产波动, 在确保药物质量的同时, 通过全流程成本控制加速产品上市。如今, QbD已成为制药行业工艺和产品设计与优化的强制性要求。
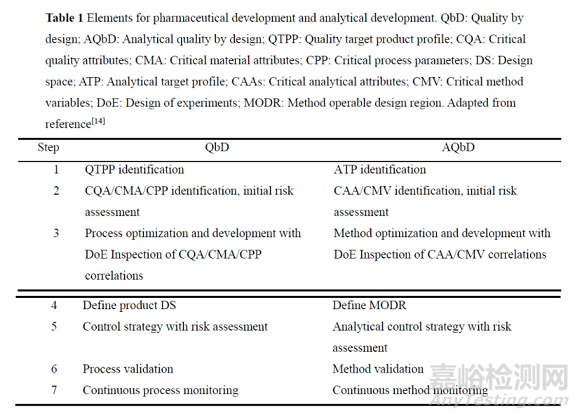
“分析质量源于设计”(analytical quality by design, AQbD)是QbD在分析方法开发中的延伸应用, 旨在建立稳健的分析方法, 有效防控超趋势(out of trend, OOT)、失控(out of control, OOC)或不合格(out of specification, OOS)等质量偏差事件, 通过在方法参数的可操作设计区域(method operable design region, MODR)内实现参数变化的监管灵活性, 从而降低分析成本。AQbD在分析方法开发中的应用与QbD在药物研发中的应用高度相似, 两者涉及的关键要素可相互映射(表1)。
近年来, 随着FDA批准的一些新药展示了AQbD在分析方法开发中的重要性和应用价值, AQbD在分析方法开发中的推广得到了进一步推动。此外, 美国药典(US Pharmacopoeia, USP)于2022年5月发布了新通则<1220>《分析方法生命周期》, 英国药典(British Pharmacopoeia, BP)也在2022版补充章节“分析质量源于设计在药典方法中的应用”中指出, 基于AQbD开发的方法, 可在经验证的范围内调整方法参数而无需重新验证, 从而为分析方法变更提供了更大的灵活性。与此同时, ICH发布了新指导原则《分析方法开发》(Q14)和《分析方法验证》(Q2(R2))并于2023年11月正式生效。这些指南均涉及AQbD框架的制定, 进一步推动了其在制药行业中的应用。在当前形势下, 结合AQbD理念开发高效、高质量的分析方法已成为制药行业的重要趋势。值得一提的是, Beg等撰写的《分析质量源于设计手册》系统地梳理了AQbD术语, 并提供了实际应用案例, 为分析人员提供了宝贵的参考。
鉴于AQbD在生物药物分析中的技术革新价值及国际监管推动趋势, 本综述基于ICH系列质量指南的框架, 结合USP、BP相关章节及最新文献报道, 系统阐述了AQbD的工作流程, 总结了AQbD理念在生物药物分析方法开发中的应用现状, 并对该领域实施AQbD所带来的机遇与挑战提出了见解与展望。
1AQbD工作流程
传统上, 分析方法开发通常采用单因素法(one factor at a time, OFAT), 即一次改变一个因素。基于该方法开发的分析方法存在实验参数耐用性范围较窄的问题, 可能导致方法转移时需要重复验证, 进而增加开发成本; 同时, 为实现同等的准确度和精密度, 该类方法往往需要开展更多的试验, 且无法评估各因素间的交互作用。相比之下, AQbD从产品质量出发, 基于科学认知将风险评估(risk assessment, RA)与方法选择相结合, 采用多因素实验设计(design of experiments, DoE), 在方法参数与预期结果之间建立联系, 最终开发出高度稳健且成本效益高的分析方法。AQbD可减少OOT、OOC及OOS结果, 实现监管灵活性, 现已成为分析方法开发的首选策略。图1展示了AQbD的典型工作流程及关键要素。
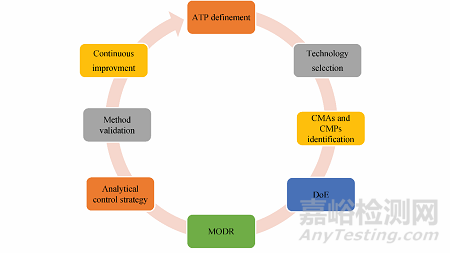
Figure 1 Typical workflow and key elements of AQbD
1.1 ATP制定
AQbD的工作流程始于分析目标概况(analytical target profile, ATP)的制定。ATP类似于产品的质量目标概况(quality target product profile, QTPP), 明确了分析方法的预期目的、关键质量属性及方法性能标准, 但不涉及具体技术或操作模式。作为AQbD的基石, ATP指导分析技术的选择, 并促进方法的持续监测与改进。在分析方法生命周期中, 方法的开发、验证及监测均以ATP为基准, 确保方法始终符合预期目的。
1.2 技术选择
确定ATP后, 下一步即是根据ATP需求, 基于先验知识或预实验选择合适的分析技术。此时需考虑一系列问题, 如: 分析方法的目的是什么? 性能要求如何? 待测样本有何特点? 实验室设备及操作条件是否满足? 方法复杂性如何? 是否需要特殊培训? 检测时间和成本如何控制? 对于这些问题的思考有助于分析人员准确理解拟开发方法的需求, 从而选择最合适的分析技术。总体而言, 应综合考虑方法的科学性、实用性和经济性, 选择适用于检测目的的分析技术; 也可使用决策树、流程图或决策矩阵等工具比较不同分析技术的优劣。
1.3 基于风险评估确定CMAs和CMPs
选定分析方法后, AQbD将聚焦于方法开发, 全面评估仪器配置、样品特性、样品制备、环境条件及方法参数等与变异性相关的风险。其主要目的是通过风险评估确定关键方法属性(critical method attributes, CMAs)和关键方法参数(critical method parameters, CMPs)。CMAs可直接衡量分析方法的变异性或准确度, 如以RSD评价精密度或回收率评价准确性; 也可以是与系统相关的属性指标, 如色谱法中的分离度、拖尾因子和信噪比等。这些指标需控制在适当范围内, 以确保数据质量。CMPs则是可能影响CMAs的潜在变量, 涵盖样品制备、分析测量和数据分析等环节。
风险评估需遵循ICH Q9指南, 分为风险识别、风险分析和风险评估三个步骤。常用的风险管理工具包括Ishikawa图(鱼骨图或因果图)和失败模式效应分析(failure mode and effect analysis, FMEA), 前者常用于识别CMPs和CMAs, 后者常用于对风险因素进行优先排序。通过FMEA, 根据各风险因素影响结果的严重程度(severity, S)、产生影响的概率(probability, P)和产生影响后检测的难易程度(detection, D), 计算风险优先指数(risk priority number, RPN), 对各因素进行打分和排序。其他工具还包括比较矩阵(comparison matrix, CM)、故障树分析(fault tree analysis, FTA)和危害分析与关键控制点(hazard analysis and critical control point, HACCP)等。
在风险评估过程中, 重点是对识别出的CMPs进行分类: 哪些参数可以固定或通过小范围调整控制; 哪些参数难以预测或控制; 哪些参数需通过实验研究确定其对CMAs的影响及可接受范围。因此, 风险评估的重要产出之一是根据FMEA的RPN打分和排序制定评价策略, 以确定需通过DoE进一步研究的CMPs, 如将超过某一RPN阈值或RPN值位于前30%的高风险参数纳入DoE研究范围, 作为DoE考察的核心变量。
1.4 DoE实验设计
DoE是一种系统的化学计量学工具, 专门用于方法开发与优化, 它通过将方法参数(输入因子, Xn)与输出响应(Yn)相关联, 帮助分析人员获取过程相关信息, 并构建数学模型以反映参数与响应之间的因果关系。根据实验目的, DoE设计通常分为筛选设计和优化设计(响应曲面设计)两大类。此外, 多种专业软件可用于DoE设计和统计建模, 如JMP、MATLAB、Fusion、Drylab、Design-Expert和MODDE等。
1.4.1 筛选设计
筛选设计能够在较少实验次数下研究大量输入因子, 识别关键因子以纳入优化实验, 并可同时评估定性和定量参数对CMAs的影响, 由于具备显著的经济效益优势而被广泛应用。通过筛选设计, 不仅能确定不影响CMAs的参数及其工作条件, 还可获得部分参数的最优值, 从而减少优化阶段需研究的CMPs数量, 降低实验次数。此外, 基于筛选实验获得的参数范围, 可进一步调整以获得最佳结果。
在筛选阶段, 通常采用两水平设计, 以较少的实验次数研究尽可能多的参数。其中Plackett-Burman设计(PBD)和部分因子设计(fractional factorial design, FFD)为最常用的方法, 尤其适用于因子数量≥5的情况。若根据现有信息能合理规划后续优化设计, 有时也可基于先验知识或初步单变量实验直接决策, 而不必开展筛选研究。
1.4.2 优化设计
优化阶段通常采用响应曲面方法(response surface methodology, RSM)来评估CMPs对CMAs的主要相互作用和二次效应。与筛选阶段相比, 优化阶段旨在评估模型是否存在曲率, 因此每个参数需至少研究3个水平, 实验次数相应增加。该阶段一般侧重于研究连续CMPs对CMAs的影响, 例如色谱法中的梯度条件、流速、缓冲液浓度和pH值等。所考察的CMPs可基于筛选试验结果或直接从风险评估中选择。
在优化阶段, 常用的对称设计包括全因子设计(full factorial design, FD)、中心组合设计(central composite design, CCD)、Box-Behnken设计(Box-Behnken design, BBD)、田口设计(Taguchi design, TD)和Doehlert设计(Doehlert design, DD)。当需要研究的CMPs数量较多时, 可采用D-最优设计(D-optimal design, DoD)以减少实验次数。此外, I-最优、G-最优和A-最优设计也可用于特定模型的优化, 这些设计可根据模型主效应或系统复杂性包含二次项, 并可用于筛选目的。优化设计还包括混料设计(mixture design, MD)以及混料–过程变量设计(mixture process variable, MPV)。
1.5 MODR
根据ICH指南Q8, 分析方法的MODR等同于产品开发中的“设计空间”(design space, DS)。ICH指南Q14将MODR定义为“由两个或多个分析方法参数的组合范围组成, 在这些参数范围内, 分析方法产生的结果始终符合ATP规定的目标”。因此, MODR可被视为分析方法的稳健性区域, 在MODR范围内调整参数将不被视为方法变更, 且在每个点上以特定概率水平保证方法性能的质量。MODR越大, 方法满足CMAs要求的稳健性越强。计算MODR时, 需考虑模型参数的不确定性及满足CMAs要求的概率等因素, 常用工具包括蒙特卡洛模拟、贝叶斯建模和自举法等。
1.6 分析方法控制策略
为保证分析方法的性能和质量, 制定合适的控制策略至关重要。“控制策略”的概念首次出现在ICH Q8(R2)中, 并在ICH Q10和Q11中进一步发展, 随后在ICH Q14“分析方法开发”中扩展至分析方法领域。分析方法控制策略(analytical control strategy, ACS)包括方法描述、待控制的分析参数及系统适用性测试(system suitability test, SST)。分析方法描述应涵盖分析测试的所有步骤, 如标准物质、样品和试剂的准备, 仪器使用, 标准曲线生成, 结果计算公式及其他必要步骤。
与传统方法的ACS相比, AQbD下的ACS可能没有显著差异, 但基于AQbD的ACS在分析方法参数的操作范围内提供了更大的灵活性, 允许参数在允许范围内波动。此外, 由于ACS基于ATP、DoE实验数据和MODR制定, 它在分析方法性能与目的之间建立了更强的联系。ACS应在方法验证前确定, 并在验证完成后加以确认。
1.7 方法验证
一旦建立ACS并将其纳入分析方法生命周期, 下一步即可开展方法性能验证, 以确认方法符合ATP的要求。验证的目的是识别不同实验室使用该方法时可能引入的变异来源, 并加深对方法的整体理解, 以确保其始终符合ATP的规定。为此, 需制定验证方案以确认方法性能, 并遵循ICH Q2等相关验证指南。
1.8 持续改进
经验证的方法投入日常使用后, 作为持续性能维护的一部分, 应建立持续收集、评估和分析与分析方法性能相关的信息和数据的程序。通过性能监控系统、纠正预防系统及变更管理系统等工具, 实现对分析方法的持续监测与改进。例如, 预先设定OOT标准, 使用统计过程控制(statistical process control, SPC)图或其他工具跟踪系统适用性数据或方法相关调查。这种持续监测使分析人员能够检测、识别并解决方法的异常; 当出现不符合时, 及时记录并采取行动。此外, 可将风险管理和风险沟通工具嵌入工作流程, 必要时基于风险评估结果实施部分或完整的再验证。
2AQbD在生物药物分析方法开发中的应用现状
AQbD是一种系统的方法论, 旨在确保“在合适的时间使用合适的方法”, 通过了解和控制变异源, 并在MODR范围内工作, 以确保监管的灵活性。近年来, AQbD在化学药、中药等领域的分析方法开发中已广泛应用, 涉及多种理化分析技术, 包括液相色谱法(liquid chromatography, LC)、紫外–可见分光光度法(ultraviolet-visible spectrophotometry, UV-Vis)、毛细管电泳法(capillary electrophoresis, CE)、超临界流体色谱法(supercritical fluid chromatography, SFC)、高效薄层色谱法(high-performance thin-layer chromatography, HPTLC)以及一些分析联用技术, 如UPLC-MS、LC MS/MS和GC-MS/MS等。
随着对AQbD理念理解的深入, 其在生物药物分析方法开发中的应用也逐渐推广。尽管AQbD在不同药物领域的应用流程相似, 但由于生物药物的复杂性, 其分析方法开发仍面临挑战。因此, 实际应用中需根据方法特点和需求保持灵活性。本节将通过不同的生物药物分析方法开发实例, 展示AQbD在该领域的具体应用场景, 以期为生物药物分析方法开发提供参考。
2.1 液相色谱法(LC)
根据文献检索, LC作为分离技术的“金标准”, 不仅在化学小分子领域是最常见的基于AQbD理念开发和优化的分析技术, 在生物药物领域也得到广泛应用。
Nompari等基于AQbD理念开发了一种快速、选择性好、灵敏度高的超高效液相色谱检测方法(ultra-high-performance liquid chromatography, UHPLC), 用于检测乙型脑膜炎疫苗Bexsero上清中的抗原含量。Bexsero是首个获批用于2月龄及以上个体主动免疫的疫苗, 用于预防B群脑膜炎奈瑟菌引起的侵袭性疾病, 其未吸附抗原含量是关键质量属性。该研究以准确定量Bexsero的5个活性蛋白成分并实现基线分离为目标, 选择了RP-UHPLC作为分析技术。研究以奈瑟菌肝素结合抗原的容量因子、抗原峰分离度和峰面积作为CMAs, 基于Ishikawa图评估识别可能影响CMAs的CMPs, 并通过预实验优化了检测器类型、样品表面活性剂等CMPs。在实验设计阶段, 采用两轮非对称筛选矩阵和CCD, 系统评估了样品小瓶类型、浓度、进样量、色谱柱类型等因素对CMAs的影响, 并通过RSM和蒙特卡洛模拟确定了CMPs的MODR。最终优化的条件使抗原在5min内实现完全分离。在方法验证阶段, 研究人员通过全因子设计进行耐用性测试, 并按照ICH Q2指南开展验证, 结果表明方法具备良好的选择性、线性、准确度(回收率90.9%~115.9%)和精密度(重复性RSD为1.7%, 中间精密度RSD为2.8%~8.4%), 且定量限低至0.5μg·mL-1, 可用于Bexsero疫苗的常规分析。此外, 还考察了分析人员、色谱柱批次、样品制备等日常使用中易变因素对结果的影响, 进一步证实了方法的稳健性。该研究首次将AQbD应用于疫苗质量控制, 展示了其在复杂生物药物分析方法开发中的系统性和科学性, 为类似产品的质量控制提供了可借鉴的开发框架。
Moineau等报道了应用AQbD建立了一种高效阴离子交换色谱–脉冲安培检测法(high-performance anion-exchange chromatography with pulsed ampero metric detection, HPAEC-PAD), 用于定量市售六价液体疫苗中非吸附多糖—聚核糖核糖醇磷酸酯(polyribosyl ribitol phosphate, PRP)的含量及其去聚合PRP的百分比。HPAEC-PAD是一种已建立的多糖变体检测方法, 其分析步骤包括低速离心提取非吸附PRP、超速离心分离去聚合多糖与原生共轭多糖, 随后在碱性条件下将PRP水解为二糖并进行HPAEC-PAD分析。研究中, AQbD用于优化超速离心和色谱分离阶段的关键实验参数。首先, 基于欧洲药典要求和液相色谱标准, 制定了ATP。同时, 根据放行标准和先前色谱法相关的历史验证数据, 明确了方法的专属性、范围、准确度、精密度和定量限等性能指标的可接受标准。随后, 采用FMEA对可能影响方法精度和准确性的CMPs进行风险评估, 识别了7个高风险CMPs, 包括超速离心的速度、温度、时间、超速离心后在上清液回收之前的保持时间以及色谱分析中流动相的氦气压力、存储时间和自动进样器中的样品存储时间。接下来, 采用Ishikawa图的方式展示了上述7个参数对分析方法精度的影响。这与多数文献中风险评估先采用Ishikawa图工具罗列所有可能影响CMAs的CMPs, 再采用FMEA进行打分排序的方式略有不同; 而且, 该研究中FMEA评分与常规风险评估也不同, 未考虑可检测性, 仅基于严重性和发生概率计算RPN。这可能是因为拟优化的方法已获批准, 研究人员对其有充分理解, 因此风险评估策略根据实际情况作了调整。在实验设计阶段, 采用L18田口正交设计研究了7个CMPs对结果的影响, 并通过18轮实验确定了每个CMP的MODR。最终, 基于对方法的理解和耐用性研究, 建立了ACS。该研究展示了AQbD在优化已批准分析方法中的应用, 显著提升了方法的稳健性和可操作性, 为复杂疫苗产品的质量控制提供了有力支持。同时, 与传统方法相比, 基于AQbD优化的方法参数, 未来在定义的MODR范围内变动时, 都将被接受和认可, 这为变更管理提供了更大的灵活性。
2.2 酶联免疫吸附法(ELISA)
ELISA作为最常用的免疫化学方法之一, 广泛用于蛋白疫苗抗原含量、体外效价或蛋白药物结合活性的测定。以下案例展示了如何在免疫分析方法开发中实施AQbD。
Yarovoi等将AQbD应用于四价疫苗候选物体外相对效力测定放行方法的开发。研究首先制定了ATP, 明确了方法的用途、开发周期及性能要求。基于ATP对精密度、耐用性、样品类型和检测通量的要求, 选择ELISA作为开发技术。随后, 使用Ishikawa图识别可能影响方法性能的因素, 并通过因果矩阵对其进行风险排序, 以确定需优化或控制的关键因素。在风险评估中, 研究团队采用FMEA计算RPN, 综合考虑严重性、发生概率和可检测性。通过棋盘滴定法初步探究了实验条件, 并根据实验结果确定因子范围, 开展后续DoE研究。DoE设计采用Design Expert软件完成, 筛选阶段使用FFD, 优化阶段采用五水平半区CCD。基于筛选结果, 将碱性磷酸酶偶联抗体和底物显色时间等参数控制在最优水平, 并对捕获和检测条件进行优化。最终, 通过第二轮CCD确认最优条件, 并采用完全巢式方差分析和Minitab软件评价方法的精密度。该研究系统展示了AQbD在ELISA方法开发中的应用, 通过风险评估和实验设计优化关键参数, 显著提升了方法的精密度和耐用性, 为疫苗效力测定提供了可靠的分析工具。
Han等基于AQbD理念开发了一种通用双抗体夹心ELISA法, 用于检测严重急性呼吸综合征冠状病毒2型(severe acute respiratory syndrome coronavirus2, SARS-CoV-2)蛋白亚单位疫苗的抗原含量。研究中, 采用猪抗SARS-CoV-2蛋白受体结合区域(receptor-binding domain, RBD)多克隆抗体作为包被抗体, 辣根过氧化物酶(horseradish peroxidase, HRP)标记的单克隆抗体20D8作为检测抗体。通过风险评估, 研究确定了包被抗体和酶标抗体浓度、抗原抗体孵育及显色温度、抗原抗体孵育时间及显色培养时间等CMPs, 并以信噪比作为CMA。采用JMP软件进行定制化实验设计, 并通过蒙特卡洛模拟确定了各参数的MODR。方法验证指标通过BMV®软件计算, 抗原含量则采用Biostat®软件中的量反应平行线法进行定量分析。经验证, 该方法具有足够的特异性和准确度, 精密度≥90%, 在70%~143%的质量标准范围内, 方法能力指数>0.96, 且误判概率<0.39%; 另外, 可检测5个不同厂家的SARS-CoV-2蛋白亚单位疫苗抗原, 展示了其广泛的适用性。该研究开发的通用ELISA方法不仅解决了SARS-CoV-2疫苗抗原含量检测的技术瓶颈, 还为已上市、紧急使用及研发中的疫苗提供了高效、可靠的分析工具, 体现了AQbD在生物药物分析方法开发中的实际应用价值。
在另一项研究中, Rodríguez等基于AQbD工具开发了一种简便可靠的间接ELISA方法, 用于检测针对重组SARS-CoV-2蛋白RBD的人IgG。研究中, 使用HEK293细胞生产和纯化的SARS-CoV-2蛋白RBD作为捕获抗原, 兔抗人IgG-HRP作为检测抗体。以提高阳性血清与阴性血清的信号比为目标, 研究对包被抗原用量和兔抗人IgG-HRP稀释度进行了三水平全因子设计, 包含4个中心点, 共完成13次实验。采用Stat-Ease Design-Expert软件分析数据, 并通过分层多项式模型拟合信号比。在响应优化阶段, 应用满意度函数确定了最佳条件, 并进行了验证。通过Cohen's kappa统计量评估, 该方法与参考方法RT-PCR的一致性达到0.92, 表明其具有较高的可靠性。该研究开发的间接ELISA方法操作简便、结果可靠, 不仅可用于检测病毒暴露, 还具备评估保护性免疫的潜力, 展示了AQbD在高通量免疫分析方法开发中的高效性和实用性。
2.3 毛细管电泳法(CE)
CE系基于电场中电荷分子的差异迁移进行分离, 目前已成功用于生物药物活性成分及相关变异体的检测。
van Tricht等基于AQbD开发了一种快速稳健的CE方法, 用于定量生产全工艺段样品中的完整腺病毒颗粒。传统方法如定量聚合酶链式反应(quantitative polymerase chain reaction, qPCR)和阴离子交换HPLC(anion exchange HPLC, AEX-HPLC)分别存在耗时长、成本高、重复性差或回收率低等问题, 难以满足全工艺段样品的定量需求。因此, 该研究以开发一种可在一天内完成分析, 且能对0.5×1011~1.5×1011·mL-1的完整腺病毒颗粒实现准确精密定量作为ATP, 同时还期望建立的方法可用于上游工艺(upstream processing, USP)和下游工艺(downstream processing, DSP)样品的定量分析。CE因其对化学小分子、多肽、蛋白质及颗粒的高效分辨能力而被选为替代方法。研究中, 从USP和DSP中选取具有代表性的一组样本, 用于优化完整腺病毒颗粒与其他基质成分的分离。通过全因子设计, 优化了Tris浓度、N-三(羟甲基)甲基甘氨酸浓度、分离电压和毛细管有效长度4个CMPs, 并采用JMP统计软件确定了各参数的MODR。方法开发完成后, 研究人员选取了裂解收获液、澄清收获液、阴离子交换纯化液、超滤液和原液等5个代表性样品进行验证, 并与传统方法结果进行比较。结果显示, 基于AQbD开发的CE方法能够精确定量含有细胞碎片、裂解液、宿主细胞蛋白、DNA、盐和去污剂等复杂基质的腺病毒样品, 加样回收率为95%~110%, 中间精密度RSD为7.8%(n=18), 重复性RSD为2.1%~4.8%(n=18), 显著优于传统方法(qPCR和AEX-HPLC重复性RSD一般分别为10%和25%)。该研究开发的CE方法不仅解决了腺病毒颗粒定量中的技术难题, 还为疫苗生产过程中的质量控制提供了高效、可靠的分析工具, 展示了AQbD在复杂生物样品分析方法开发中的优势。
另外, Simeoni等报道了采用AQbD理念开发了还原型毛细管电泳法(reduced capillary electrophoresis sodium dodecyl sulfate, rCE-SDS), 以取代SDS-聚丙烯酰胺凝胶电泳(SDS-polyacrylamide gel electrophoresis, SDS-PAGE)作为商品化单克隆抗体产品的放行和稳定性测试方法, 展示了如何通过AQbD对过时技术进行更新。研究中, 首先明确了方法的预期用途: 在还原条件下分析和定量IgG1X变体, 以替代SDS-PAGE。尽管RP-HPLC和rCE-SDS均可满足ATP要求, 但rCE-SDS对IgG1X片段的分离度更优, 且符合USP通则“重组治疗性单克隆抗体分析方法”的推荐, 因此被选为目标方法。通过Ishikawa图进行风险评估, 确定了重链峰拖尾因子、非糖基化重链峰分离度等CMAs和迁移电压、缓冲液稀释度和样品pH等CMPs。随后, 采用MODDE软件进行DoE优化设计, 确定了方法的MODR。方法开发完成后, 按ICH Q2指南进行全面验证, 并通过桥接研究比较了rCE-SDS与SDS-PAGE在检测IgG1X变体方面的一致性。结果显示, 两种方法均能指示样品稳定性, 但rCE-SDS比SDS-PAGE更灵敏, 测得的杂质总量更高, 而且对于SDS-PAGE无法定量的非糖基化重链也可进行定量, 通过建立两种方法测定结果间的转换因子补偿了灵敏度差异, 避免了历史数据的丢失, 为方法变更提供了支持。该研究展示了AQbD在方法变更中的应用, 通过系统风险评估和实验设计, 成功开发了更灵敏、更可靠的rCE-SDS方法, 为单克隆抗体产品的质量控制提供了高效工具。
2.4 其他分析技术
除上述分析技术外, AQbD工具还可应用于生物药物其他方法的开发和优化。
Yao等基于AQbD理念开发了一种UV-Vis方法, 用于测定L-天冬酰胺酶(L-ASNase)的活性, 以满足药品质量控制的需求。研究中, ATP被定义为开发一种操作简单、省时且成本低, 能够准确精密测定L-ASNase药物制剂的活性的方法。通过对Nessler法、Berthelot法、吲哚克辛法、谷氨酸脱氢酶法和天冬氨酸转氨酶法的性能特征、操作便捷性和成本进行比较, Nessler法被选为目标开发技术。研究评估了测定过程的每个步骤, 包括溶液制备、实验操作和数据处理, 确定了450nm处的吸光度值(A450nm)及其精密度作为CMAs; 而CMPs分别从Nessler方法和酶活性反应条件进行评估, 其中Nessler方法选择CKI/CHgI2、CNaOH/CHgI2、CHgI2final和反应时间作为4个CMPs, 而酶活性反应条件选择了反应温度、L-天冬酰胺浓度、缓冲液pH值和KH2PO4浓度4个对L-ASNase活性有显著影响的CMPs。最终, 该方法的MODR为Nessler法和酶活性反应条件MODR的组合。通过设置系统适用性要求, 以保证方法性能的一致性, 并建立了ACS。实验结果表明, 基于AQbD开发的UV-Vis法能够有效测定加压处理和未加压处理的L-ASNase活性。该研究展示了AQbD在酶活性测定方法开发中的应用, 通过系统风险评估和实验设计, 成功开发了一种高效、可靠的UV-Vis方法, 为L-ASNase制剂的质量控制提供了有力支持。
Pathak等基于AQbD理念开发了一种新型天然聚丙烯酰胺凝胶电泳法(native gel electrophoresis, N-PAGE), 作为分析单克隆抗体聚集体的低成本工具, 并与现行金标准分子排阻色谱法(size exclusion chromatography, SEC)进行了比较。SEC设备昂贵且存在固定相与分析物二次相互作用导致洗脱时间较长等问题, 研究提出了将N-PAGE作为正交分析方法。研究中, 首先对运行缓冲液pH值、摩尔浓度、氨基己酸浓度、凝胶缓冲液pH值、上样染料、样品浓度和运行电压等参数进行了初筛。随后, 通过两轮DoE设计优化分离胶条件和凝胶百分含量。第一轮DoE以运行缓冲液pH值、摩尔浓度、氨基己酸浓度和凝胶缓冲液摩尔浓度为CMPs, 聚集体条带数为CMA, 采用JMP软件进行实验设计。第二轮DoE采用三因素两水平全因子设计, 通过监测聚集体条带数量、扩散情况及聚集百分比误差评估分离质量。实验结果表明, N-PAGE与SEC在分析单克隆抗体聚集体方面具有相当的一致性, 且成本显著降低。该研究开发的N-PAGE方法不仅为单克隆抗体聚集体分析提供了一种低成本、高效的替代方案, 还展示了AQbD在优化电泳方法中的应用潜力, 为生物药物质量控制提供了新的工具。
Kochling等基于AQbD理念开发了一种具有通用性的平台分析方法, 用于分析蛋白质的不同关键质量属性, 展示了AQbD在平台方法开发中的潜力。研究中, 考虑到肽图分析、氧化物检测和C末端变异体分析具有相同的样品前处理步骤(还原、烷基化和脱盐), 研究人员将这三种方法的开发同步进行, 以优化资源利用。根据分析目标, 分别选择UHPLC-UV、UHPLC-MS和UHPLC-UV作为肽图、氧化物和C末端变异体拟开发的分析方法。开发过程中, 既有对三种方法共有的步骤样品前处理条件的考虑, 也有针对每种方法各自不同的检测特点特定的考虑, 其中风险评估均采用Ishikawa进行, 色谱方法开发软件采用Drylab。在肽图分析中, 研究以两个关键肽段的酶切错切量为CMA, 还原时间、还原温度、烷基化时间和烷基化温度为CMPs, 通过全因子设计和RSM确定了前处理的通用条件和MODR。在氧化物LC-MS定量分析中, 研究以待测峰与相邻峰的分离度(Rs)为CMA, 评估了去溶剂化气体流量和温度的影响。对于C末端变异体分析, 研究人员通过Drylab软件进行全因子设计模拟, 优化了色谱分离条件, 将开发时间从1~3个月缩短至1周左右。该研究通过AQbD策略成功开发了通用的平台分析方法, 显著缩短了开发时间并降低了成本。其系统性风险评估和实验设计为蛋白质多属性分析提供了高效、可靠的解决方案, 展示了AQbD在平台方法开发中的显著优势。
3总结与展望
AQbD是一种基于风险评估的系统开发策略, 旨在开发高质量的分析方法, 提升方法性能, 同时满足监管灵活性, 确保方法在稳健的MODR范围内运行, 避免方法转移过程中的故障。其核心是在方法设计和开发阶段, 聚焦于对最终产品质量有重要影响的因素的理解和控制, 包括方法选择、样品前处理、仪器性能等。通过系统研究这些因素, 可以更好地预测和控制方法性能, 从而实现高质量的药物分析。
为支持AQbD的实施, ICH、USP及BP陆续发布了相关指南。其中, ICH于2023年11月发布的Q14与Q2(R2)指南终稿, 提供了全球统一的协议框架, 并在关键要素和术语上达成一致, 以推动分析方法的全生命周期管理。根据文献调研表明, AQbD在化学小分子药物和中药分析方法开发中已广泛应用, 而生物药物由于其复杂性, 给分析方法的开发带来挑战, 使AQbD在该领域的应用尚处于起步阶段。尽管如此, 这一系统化的开发策略正逐步得到认可和推广。AQbD可有效应用于疫苗、单抗、重组蛋白药物及酶等多种生物药物领域, 既可用于全新方法的开发, 也可用于已批准方法的优化或替代。此外, 它不仅适用于高纯度原料药或制剂的活性成分及杂质分析, 还适用于中间品及强制降解产物等不同工艺阶段的产品分析。涉及的分析手段既包括色谱法、CE、LC-MS等理化分析技术, 也包括ELISA、PAGE等免疫分析方法, 甚至在平台分析方法开发中也有尝试。
从不同方法的开发和优化流程可以看出, AQbD开发策略并非固定模式, 而是基于实际开发目的和便捷性, 有选择性地涵盖重点要素。在实际应用中, AQbD强调建立分析方法的设计空间, 即在特定参数范围内确保方法的稳健性和可靠性, 通常涉及优化条件的确定及对潜在变化的敏感性评估。与传统开发方法相比, AQbD开发策略的优势在于分析方法可在全生命周期管理中进行持续改进, 即基于开发过程中的知识和经验不一定可以获得“满分”方法, 但可以通过在日常使用中的持续监测, 继续改进解决该方法仍然存在的缺点。随着各种创新生物药的迅猛发展, 基于多技术联合分析的场景越来越多, 比如CE与MS联用, 流式细胞术与MS联用, qPCR与高通量测序联用等, 由于AQbD策略可实现从“经验驱动”向“数据驱动”的模式转变, 将通过理念创新与监管协同, 为这些复杂的生物分析方法开发提供便捷和保障, 并有望将生物药分析方法开发效率提升30%~50%, 同时降低验证失败率和方法生命周期内的管理成本。当然, 为了充分利用AQbD在分析方法开发中的优势, 分析人员需具备深厚的专业知识和敏锐度, 针对不同方法的特点, 制定有针对性的开发策略, 以有效管理知识和风险, 提高数据质量并降低重新开发和验证的成本。基于开发经验, 分析人员可不断建立新的知识空间, 并将其应用于后续工作。
尽管AQbD在生物药物分析方法开发中显著提升了分析效率并减少了资源消耗, 但仍面临一些挑战。例如, 生物药物分析方法开发本身的复杂性、对设备和技术的高要求、人员培训及知识管理和风险管理的综合性等问题, 仍需在成本效益之间取得平衡。未来, 随着技术的发展和经验的积累, AQbD有望在生物药物分析领域发挥更重要的作用。通过深入研究AQbD的应用, 可以更好地理解其原理, 并为其进一步发展提供有价值的参考。

来源:Internet


