您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-08-08 18:30
摘要
单克隆抗体(monoclonal antibody, mAb)是临床应用最多的一类治疗性蛋白,虽然目前上市的mAb主要用于治疗非感染性疾病(如肿瘤或自身免疫疾病),但自新冠疫情暴发以来,国内外获批用于治疗或预防感染性疾病的单抗类药物数量正逐渐增加。世界卫生组织(WHO)于2023年3月25日发布了《针对预防和治疗感染性疾病的单抗类药物的非临床和临床评价指导原则》,中国尚未发布类似的指导原则,本文介绍WHO指导原则中预防和治疗感染性疾病的单抗类药物非临床评价相关内容,特别关注WHO的监管考虑因素、非临床评价的总体考虑以及药效学、药代动力学和毒理学研究等,以期为我国此类新药的非临床研究和评价提供参考。
关键词
世界卫生组织;感染性疾病;单抗类药物;非临床评价
单克隆抗体(monoclonal antibody, mAb)是临床应用最多的一类治疗性蛋白,而目前上市的大多数mAb都用于治疗非感染性疾病,如肿瘤或自身免疫疾病,迄今为止国内外获批用于治疗或预防感染性疾病的数量不多(表1),但自2019年新冠疫情(coronavirus disease 2019, COVID-19)暴发以来,该类产品数量正在逐渐增加。同时随着重组生物技术和蛋白质化学的进展,以及对mAb结构和功能的深入了解,重组mAb药物(如嵌合mAb、mAb片段、单结构域mAb和多特异性mAb)可能具有显著的生产、处方和临床优势,如提高产量、提高稳定性、提供替代给药途径、多抗原靶向、延长半衰期、增加生物利用度、增加功能活性和/或改变组织渗透性等,越来越受关注。由于mAb相对安全,临床起效快,且投入生产所需的时间相对较短,可为新发传染病提供及时的控制药物,同时由于其在突发公共卫生事件(如COVID-19)以及治疗慢性传染病[如获得性免疫缺陷综合征(immunodeficiency syndrome, AIDS)]中发挥的作用,mAb被认为是高度优先开发的药物。因此,世界卫生组织(World Health Organization, WHO)于2023年3月25日发布了《针对预防和治疗感染性疾病的单抗类药物的非临床和临床评价指导原则》(以下简称“指南”),经2023年3月20-24日召开的WHO生物标准化专家委员会第77次会议审议通过,并在WHO技术报告丛书中发表。

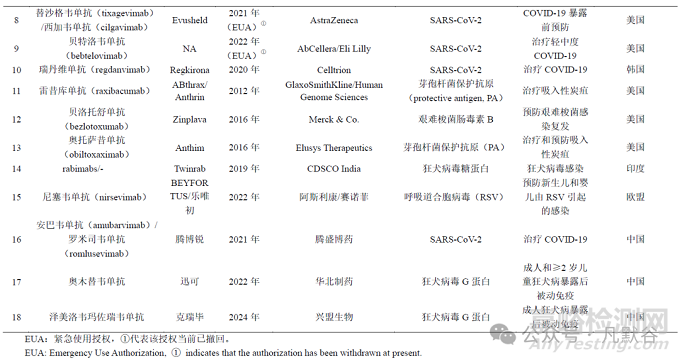
为了更好地了解用于治疗或预防感染性疾病mAb的特性及非临床研究评价要点,本文详细介绍指南中监管考虑因素、非临床评价一般考虑以及药效学(pharmacodynamics, PD)、药动学(pharmacokinetics, PK)、毒理学试验的关注要点等内容,以期为我国此类新药的非临床研究和评价提供参考。
1指南的目的和范围
WHO此前发布的与mAb相关的指导文件主要侧重于将其用作非感染性疾病的生物治疗药物,几乎没有提供关于暴露前预防(pre-exposure prophylaxis, PrEP)、暴露后预防(post exposure prophylaxis, PEP)和感染后mAb治疗的非临床或临床评价指导。该指南澄清了mAb抗感染性疾病研发和注册许可过程中的监管要求,以促进国际监管协调工作,从而改善此类产品的可及性。指南旨在为用于入侵病原体抗原或毒素暴露前和暴露后的预防或治疗人类感染性疾病的mAb进行非临床和临床评估提供指导。
指南中一般原则也适用于以预防或治疗感染为目的的靶向内源性人蛋白的mAb(如,可防止病毒进入细胞的针对细胞表面受体的mAb),但根据蛋白靶点的不同,此类产品可能需要开展额外的非临床和临床研究。免疫调节抗体靶向的是宿主免疫应答成分因子(如T细胞或细胞因子),而不是传染原本身或者毒素抗原,因此不在本指南讨论范围内。
指南适用于mAb以及基于免疫球蛋白支架的其他重组mAb相关抗原结合蛋白。这些产品包括但不限于:抗体片段(如单链可变片段和抗原结合片段(Fragment antigen-binding, Fab)片段);单结构域抗体;双特异性或多特异性抗体;经过化学修饰(如偶联)的mAb或相关抗体蛋白;为了延长半衰期、增强或降低效应功能而进行修饰的mAb(如通过序列替换、添加和/或改变糖基化)以及由多种mAb物质共同配制的终产品(抗体混合物)。该指南只有部分内容可能同时适用于小型重组mAb模拟蛋白和具有病原体特异性的血浆来源的免疫球蛋白。该指南同样不适用于使用DNA、RNA或病毒载体技术递送基因序列(给药后在体内编码生成mAb产品)的核酸平台。
关于mAb生产和质量控制的指南,参见“WHO Guidelines for the production and quality control of monoclonal antibodies and related products intended for medicinal use”。对于作为生物类似药开发的mAb产品,应参考“WHO Guidelines on evaluation of biosimilars”。
该指南的附录中还提供了一些关于mAb简化监管申报的非临床和临床方面的考虑。如果有需要,WHO将还可能针对这些产品的特定疾病监管考虑制定单独的补充指南。
2指南的一般考虑
WHO指出,最早在19世纪末人类恢复期血清和免疫动物血清被用作针对细菌和病毒感染的免疫疗法。目前来自人和马血浆的免疫球蛋白(如抗狂犬病、抗乙肝病毒和抗破伤风免疫球蛋白)仍在使用,但血清产品可能面临标准化、安全性、供应和可及性问题。与免疫抗血清和多克隆抗体相比,mAb产品提供了更可靠和更大的商业供应优势,具有更好的批间一致性和安全性,并且可设计为具有更长半衰期,具备更好特异性和功能性。
采用当前的mAb生物工程和生产技术可能快速开发出针对新发感染性疾病(无可用疫苗或治疗方法)的新产品。疫苗接种的主动免疫可能需要数周且可能需要多次接种才能出现保护作用,而给予mAb进行被动免疫可以快速直接的预防或治疗感染性疾病,因此,mAb正在成为除预防性疫苗和小分子抗微生物药物以外的用于治疗和预防感染性疾病的重要补充。
2.1 抗感染mAb的作用机制和理化特性
目前,抗感染mAb大多是全长免疫球蛋白G分子(immunoglobulin G, IgG),可中和病原体并抑制其与人体细胞受体结合直接发挥作用,通过肝脏中的库普弗细胞和窦内皮细胞的结晶片段(fragment crystallizable, Fc)受体依赖性摄取,将毒素、细菌、病毒或其他病原体从血液中清除;还可通过其Fc段介导的效应功能机制刺激免疫应答发挥作用,如抗体依赖的细胞介导的细胞毒性(antibody-dependent cellular cytotoxicity, ADCC)、抗体依赖的细胞介导的吞噬作用(antibody-dependent cellular phagocytosis, ADCP)、补体依赖的细胞毒性(complement-dependent cytotoxicity, CDC)或调理吞噬作用。因此了解mAb的作用机制对于评估其在非临床和临床研究中的活性至关重要。
除作用机制外,mAb的理化特性也很重要,如mAb分子大小、电荷改变、翻译后修饰、偶合、疏水性、聚集潜力、糖基化模式或C末端异质性。以上生化特性会显著影响mAb的半衰期、组织分布、稳定性、对酶降解的敏感性、排泄、药理作用和/或反应原性潜力。例如,对mAb的Fc区进行氨基酸工程改造可以延长半衰期,并增强或降低效应功能(如与宿主Fc受体或补体系统蛋白的相互作用)。此外,翻译后糖基化差异也会导致功能和半衰期改变。因此,每种mAb产品可能呈现出独特的理化特征,在评估过程中应予以考虑。
2.2 单克隆抗体递送
mAb的生物分布以及到达病原体感染部位的能力是产品研发过程中的重要考虑因素。mAb的理化性质、处方及给药途径都将影响药物的分布。目前大多数mAb通过静脉途径给药,通常在专门的医疗机构给药,且给药时间从30min到几小时不等。而采用高浓度mAb皮下或肌肉注射给药只需几分钟,受到越来越多的关注。另外也有开发鼻腔、吸入、口服、眼内、鞘内和皮肤给药途径。鉴于免疫球蛋白浓度、粘度、聚集和稳定性等问题,静脉和其他给药途径的mAb产品在制剂和安全性方面均有不同,因此在非临床和临床评价中需予以关注。
2.3 潜在的不良反应
在产品的研究方案中应评估mAb的两种潜在不良反应,即抗微生物耐药性的出现和抗体依赖性增强作用(antibody-dependent enhancement, ADE),并在获批上市后对其进行监测。
耐药性:与抗微生物小分子药物相似,感染性病原体也可能会对mAb产生耐药性,应在产品整个生命周期内进行监测。例如,细菌可被诱导产生抗体降解蛋白酶或可能通过自然诱变选择过程改变靶抗原,从而降低mAb的疗效。同样,病毒中多个毒株和逃逸突变体的出现可能导致形成新的变异株,从而逃避mAb的治疗。
ADE:对治疗感染性疾病的mAb(特别是当表位的功能尚不清楚时),在非临床和临床研究中都需重点关注ADE。疾病加重可能通过mAb协助完成病原体生命周期发生(如使病毒更容易进入细胞、促进靶细胞中病毒复制或促进细胞间传播),也可通过生理反应的过度增强(如补体激活)出现。
2.4 监管考虑因素
指南的非临床和临床评价部分介绍了评估mAb安全性和有效性的常规方法,可能适用于大多数已开发的产品。然而,基于特定流行病学情境开展的获益-风险评估,可能或有必要要求申请人与国家监管机构(national regulatory authority, NRA)考虑采用替代方法评估产品的安全性和有效性,以保证在紧急需要的情况下,尽可能同时满足对药物安全性和有效性的监管要求以及药物可及性。但是,必须始终考虑采取这些方法的获益风险比,并建议尽早咨询NRA。
3指南中mAb非临床评价的相关内容
WHO指出,对用于感染性疾病的mAb类药物,指南旨在补充《WHO Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology》的B部分和附录5以及《WHO guidelines on nonclinical evaluation of vaccines》,因此本指南需与上述2份指南相结合。同时,须参考《WHO Guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products(SBPs)》与《WHO Guidelines on procedures and data requirements for changes to approved biotherapeutic products》要求。此外,还应当参考国际人用药品注册技术协调会(International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, ICH)ICH S6(R1)和ICH M3(R2)以及NRA的其他相关指南。
在非临床研究中,mAb的早期发现和表征通常包括采用多种分析方法对大量mAb候选物进行筛选,以评价其在病原体或毒素中和方面的有效性并确定其可能的作用机制。虽然这些试验通常采用研究批次样品,但如果可行,应尽量使用能代表临床拟用样品的mAb产品批次进行后续的作用机制和药效学研究。如果不可行,则研究批次样品与临床拟用批次样品的理化特性数据、生物活性、稳定性和处方应具有可比性。此类研究可能包括采用表达mAb的多克隆细胞群生产的mAb产品批次进行的初步体外试验或体内试验,并以此作为分离稳定、高表达单抗产品的第一步。当使用新的或变更的生产工艺,或者对正在研发的产品或处方进行重大变更时,应证明受试物的可比性。可比性可基于生化和生物学特性(即鉴别、纯度、稳定性和效价)进行评估。在某些情况下,可能需要进行其他方面的研究,如非临床PK、PD研究和/或毒理学研究,并提供所采用方法的科学依据。
需要注意的是,关键非临床研究中使用的mAb产品批次必须能充分代表后续临床拟用样品的质量和处方。关键的非临床毒理学研究应符合药物非临床研究质量管理规范(good laboratory practice, GLP),而剂量探索研究、早期毒性研究等试验可能未遵循GLP,但应保持数据的完整性。所有动物研究都应遵循3R(“替代、减少、改进”)原则。
3.1 非临床评价的一般考虑
非临床体外和体内研究的主要目的是在开始人体研究之前确定受试物的药理学和毒理学作用,包括:(1)识别产品的功能特性,如预防疾病、减少病原体负荷、降低毒素活性、促进病原体从血液和组织中清除的能力,改善临床症状,预防或缓解体重降低,或降低感染严重程度的能力。(2)识别产品可能的毒性、毒性可逆性以及潜在不良反应。(3)确定首次人体试验(first-in-human, FIH)的安全起始剂量,并尽可能确定安全递增剂量。WHO认为,在开展非临床研究时需要考虑几个重要因素。首先,需要了解病原体的mAb靶抗原及其生物学知识,表征结合位点/表位并评估mAb对病原体的特异性和选择性。其次,需要探索与动物或人类细胞(和/或组织)的非预期交叉反应性。此外,在某些流行病期间,抗原可能会自然发生改变(即通过抗原漂移或转移),导致mAb对靶抗原的亲和力降低。因此,在将mAb投入临床研究之前,应考虑并且前瞻性地评估表位突变导致亲和力降低的可能性,并由申办者进行监测(如采用流行的和新出现的菌株的抗原进行体外测试)。
选择合适的动物种属用于评估抗感染mAb可能具有一定的挑战性,而且在不同的研究中不一定采用相同的动物种属。应提供每项试验动物种属选择的科学性依据,并应考虑所得试验数据对指导人体临床研究的适用性。应与NRA讨论动物种属的选择以及在一项研究中合并观察指标的可能性,特别是当感染动物模型尚未建立,或者当感染动物模型与人体生理学不相关或不能反映人体感染的病理学时。需要注意的是,mAb产品本身的性质也可为种属选择提供参考。尽管对于感染病原体而言,无论宿主是什么,抗感染mAb的靶抗原是独特的,但在非临床研究中,宿主对mAb结合病原体的后续反应可能因宿主种属和mAb来源种属不同而有显著差异。例如,在小鼠模型中使用人源化mAb不一定能预测相同的人源化mAb在人体中的活性或安全性。因此,了解宿主种属和mAb差异的影响对于临床前开发计划以及非临床到临床数据转化至关重要。
抗药抗体(anti-drug antibodies, ADAs)的产生具有种属特异性,动物研究中ADA的出现通常与预测mAb产品在人体中的潜在免疫原性无关。尽管如此,特别是对于mAb相关产品,在动物体内检测ADA可能有助于了解其潜在并发症,也可能有助于解释动物毒性研究数据。例如,ADA的形成会增加mAb的清除并影响其PK和/或毒代动力学(toxicokinetics,TK),从而降低其药理学和/或毒理学作用。ADA的诱导还可导致其他药理学和/或毒理学变化,包括出现新的毒性反应。因此,应考虑ADA形成对PK和TK的此类影响。
此外,还应考虑mAb的作用机制涉及次级反应(如ADCC、ADCP或CDC)的情况,上述反应可能因抗体Fc和动物模型Fc受体的不同而产生很大差异。在解释动物研究中的暴露-反应关系、PK参数和组织毒性时,应考虑这些药理学特性以及它们是否具有种属特异性。此外,还必须考虑动物感染模型与人类感染的相似程度。
在所有动物研究中,对攻毒用病原体毒株进行测序、表征和标准化并明确其感染剂量非常重要。在病原体传代可能导致变异株(如SARS-CoV-2)产生的情况下,使用在明确且标准化传代水平下的攻毒物质至关重要。此外,对给予mAb后仍因感染而死亡的动物体内分离出的病原体进行基因分型,有助于分析此类感染的易感性是否与病原体的抗原漂变或转变相关。
3.2 药效学和生物活性
3.2.1 体外药效学试验
可以采用体外试验评估mAb的生物活性。应采用多个浓度受试药开展体外药理学研究,对于mAb片段或免疫偶联物,应采用该形式的受试物开展试验。若更新更合适的分析技术已经通过验证,则应予以采用。
体外机制研究一般包括对结合位点的表征,对外源性靶标、感染性微生物或细菌毒素的结合亲和力,对病原体灭活/破坏机制研究[如杀菌、调理吞噬或中和活性(包括对变异株的影响)]以及对mAb的功能活性测定。结构生物学方法也可用于在原子水平上绘制mAb抗原复合物图谱。体外研究也可能有助于评估以下因素的影响:(a)抗原变异,如通过遗传漂变或转移自然发生;(b)细菌被膜转换;(c)病原体逃逸突变。这些抗原变异株可能是在实验室分离的,也可能来自临床分离株。申办者还应考虑与其他已上市抗体/药物产生交叉耐药的可能性。
(1)细胞培养试验
细胞培养模型对于候选药物的早期筛选、mAb抗体对目标病原体作用的评估以及mAb作用机制的探索可能是非常有用的工具。细胞培养试验是体外评估mAb中和活性和抗体效应功能(如ADCC、ADCP或调理作用)的重要组成部分。然而,某些时候可能尚未建立适用于所有传染性病原体的细胞培养系统,特别是在疾病大流行的早期阶段或当病原体对细胞培养方法或环境不耐受时。若建立了细胞培养模型,应确保环境条件可维持mAb的正常功能,并尽量减少分析试剂的干扰。在细胞培养模型中使用来自不同种属的组织或细胞也可能有助于了解PD研究中的最相关的动物模型。对于联合配制的mAb,应测试每种单独mAb的中和活性,并明确不同抗体之间的任何潜在协同或拮抗作用。
(2)组织交叉反应试验
mAb与非靶组织结合可能会引起严重后果,尤其是在使用某些免疫偶联物时。因此,通常应在FIH研究之前进行组织交叉反应研究,以检测任何可能的非靶组织结合或其他交叉反应。应使用一组冷冻组织或有代表性的细胞培养物来确定试验用mAb与人体组织的非预期反应。由于检测交叉反应的能力可能取决于mAb的浓度,因此应采用mAb多个浓度进行试验。对人体组织样本的相关要求,应咨询NRA。同样,应与NRA讨论使用经过验证的细胞和/或蛋白质微阵列分析法评估与人类蛋白质的脱靶反应性的可能性。当检测到交叉反应信号时,应将研究扩展到更多组织。虽然使用动物组织可能有助于解释动物研究中的一些结果,但不推荐采用一整套完整的动物组织进行组织交叉反应试验。
3.2.2 体内药效学试验
体内药效学研究对于了解mAb在生物系统中的生物活性非常重要。由于动物药效学研究也用于估算FIH剂量,因此应尽可能进行此类研究。但是,由于对mAb药效学研究的要求可能因国家/地区而异,并且若具备足够的相关数据和经验,监管机构可能允许采用体外和/或模拟试验作为替代方法,因此针对这一点,在mAb开发过程中应尽早与NRA讨论。药效学研究应当采用确保mAb能够作用于目标感染原的试验方法。由于在动物模型中进行经典PK/PD研究可能与人体的相关性有限。对于大多数病原体,可以参考在疾病及其预防工作中对相关试验方法积累的丰富知识和经验。此外,现有的关于自然免疫和/或疫苗诱导的免疫相关知识也可能为正在研发的mAb的非临床评估提供额外的参考。
若存在感染动物模型时,应尝试研究mAb药效学作用的剂量依赖性。使用较宽的剂量范围(包括高剂量)有助于更好的预测治疗指数。当最终产品采用两种或更多的mAb,可仅对最终产品进行动物体内评估,在体外对每种mAb及最终产品的药效学作用进行评价。
对于抗病原体活性的概念验证研究,应优先选择动物感染与人类感染相似的模型开展试验,考虑所选动物模型的感染与人类感染和疾病的相似程度。由于该指导原则包含的mAb和感染性疾病种类繁多,应根据具体情况确定动物种属的选择,并提供科学依据来证明所选模型的合理性。此外,还应评估候选mAb产品,以制定最有效的治疗方案。
如果不存在感染动物模型,或者由于供应或伦理原因无法使用,则需要论证替代方法的合理性并咨询NRA。mAb药效作用的支持性证据也可能来自恢复期的人体血清,如血清抗体可以识别类似的抗原并中和或去除感染原。
3.2.3 安全药理学试验
安全药理学研究的目的是研究候选mAb产品对机体重要功能和主要生理系统的影响,通常包括心血管、呼吸和中枢神经系统。但根据ICH指南,若能提供豁免这些研究的合理理由,安全药理学研究也可能不是必需的。可以考虑将心血管、呼吸和中枢神经系统指标的研究纳入毒性研究中。
mAb的组织分布可能受多种理化性质(例如,分子大小和糖基化)以及来源或处方的影响。因此,在评估产品对机体重要功能和生理系统的影响时,应考虑这些因素。
3.3 药代动力学和毒代动力学
进行PK和TK研究是为了了解药物在动物体内的暴露情况,以便进行动物与人体外推,并根据暴露情况预测临床试验的安全范围。更多信息可参见指南《WHO Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology》第B.3节。尽管PK和TK评估可以整合到更广泛的药理学和/或毒理学研究中,但当mAb直接作用于感染原而缺乏相关的动物模型时,对PK和/或TK数据的解释可能存在一定的局限性。
PK和TK的研究设计,以及对PK和TK数据的解释还应考虑mAb或免疫偶联物的性质、稳定性、结合血清蛋白的能力、动物是否存在感染和/或受体动物模型中的靶抗原表达水平,以及给药途径。
3.3.1 检测方法
选择PK和TK研究方法需要case-by-case,并且应提供科学依据。动物试验和人体试验的检测方法最好一致,并且应使用适合动物模型和种属的经验证的方法。应研究并考虑血浆/血清中的血浆结合蛋白和/或抗体对所选检测方法性能的可能影响。针对特定产品的检测方法应具备以下特征:可表征药理学/毒理学或PK特点;能代表和/或预测临床情况;可广泛表征所有功能特点(如半衰期);适用于所测定的产品并且经过充分论证。
3.3.2 其他考虑
吸收:经静脉给药的mAb不需要进行吸收研究。但对于其他给药途径(如肌肉注射或皮下注射),在开始人体I期临床研究前,应评估药物吸收和生物利用度。
分布:应酌情研究并考虑mAb的物理化学和动力学特性,以及不同给药途径药物的分布的差异。尽管mAb最初可能分布于血管内,但之后可能会由于各种因素(包括血流灌注和主动转运)而分布到血管外组织。
代谢:单抗通常不需要进行传统生物转化研究,但偶联mAb需要了解偶联分子去偶联后的代谢途径。
消除:在临床研究前应获得相关动物模型中消除/清除的信息,以便根据暴露量和给药剂量预测安全范围。对于免疫偶联物,还应提供偶联分子的消除信息。
3.4 毒理学研究
由于该指南涵盖的mAb和感染性疾病范围很广,应根据具体情况选择动物模型和毒理学研究,并加以论证。当使用疾病动物模型进行概念验证研究时,可以纳入毒理学评估,以提供更多有关潜在靶标相关毒性信息。若不可行,应考虑采取合适的风险控制措施,并就风险控制措施与NRA进行讨论。
对于表现出与人体组织脱靶结合和/或在动物研究中产生毒性的mAb,可能需要进行额外的毒理学试验。
该指南也提及了一篇已发表的关于治疗性抗体非临床安全性评价的综述,强调在设计非临床研究时的重要考虑因素、需要开展的非临床安全性研究类型以及与临床试验相关的非临床安全性研究的一般时间表。
3.4.1 毒理学试验一般考虑
应开展一项多剂量的短期重复给药毒性试验。若mAb在预防性治疗或感染过程中需多次给药,毒理学试验的给药方案应当反映在最差临床情况下的给药方案。试验恢复期应能反映mAb消除时间(如5个半衰期)。若仅进行单次给药毒性试验,应提供依据(如对于半衰期长的mAb),还应说明动物种属选择的合理性。
应与NRA讨论毒理学试验的要求。在理想情况下,应选择健康动物进行试验,以在无疾病状态下更清楚地解释药物的毒性,也可以模拟健康受试者当出于预防目的而给予mAb的情况。应采用雄性和雌性动物开展试验,此外,动物的发育阶段应当能反映目标人群(如年轻人、中年人、老年人)最敏感的阶段。试验动物数量根据所选择的研究动物是啮齿类动物或是非啮齿类动物而异。给药途径应能反映其临床研究拟用给药途径。
当两种或多种mAb共同配制或以其他方式开发用于联合使用时,应对联合mAb进行试验。观察到任何不良反应,都可能需对每种mAb单独进行进一步评估。对于免疫偶联物产品,应对免疫偶联物进行非临床安全性研究。此外,应评估偶联物分子(即“有效荷载”)的安全性且其毒性可被接受;否则,可能需要根据相关指南进行进一步研究。
ADA的产生可能会使动物毒理学研究以及对试验结果的解释变得复杂,若发生免疫介导反应,则应当考虑ADA因素。若根据重复给药毒性研究预测对人体的潜在影响时,应考虑ADA的形成以及相关的免疫原性问题,并应与NRA进行讨论。对于人类感染性疾病,临床上可能并不需要长期反复用药,因此在临床上诱导抗mAb免疫应答的风险可能会有所降低。
应根据已建立的方法评估mAb的局部耐受性(如对红斑/焦痂和水肿的评估)。如果可行,可在毒性研究中评估产品潜在的局部不良反应,而不需进行单独的局部耐受性试验。
3.4.2 遗传毒性和致癌性
遗传毒性和致癌性试验通常不适用于mAb。但是,免疫偶联物可能需要开展该研究,应根据具体情况考虑。
3.4.3 生殖毒性
对靶向传染性病原体(即非人类抗原)的mAb,可能不需要进行生殖毒性研究,但这一要求可能因国家/地区而异,应提前与NRA讨论,因为各个国家的指南可能与涉及生殖毒性试验的其他指南[如ICH S6(R1)]一致或不一致。NRA可能要求对拟用于育龄妇女的mAb(特别是该产品是免疫偶联物或临床经验很少的非传统mAb蛋白)进行生殖毒性研究。
在开展研究时,具体的研究设计和给药方案可能会根据不同的情况如种属特异性、免疫原性、生物活性和/或长消除半衰期等而改变。在分析结果时,还应考虑妊娠期间胚胎-胎仔暴露的种属特异性。通常,高分子量蛋白(>5kDa)不能通过简单的扩散透过胎盘,但对于分子量高达150kDa的抗体,存在一种涉及新生儿Fc受体的特定转运机制,该受体表达决定胎儿暴露量且存在种属差异。在人类和非人灵长类动物器官发生期,IgG跨胎盘转运较低,在妊娠中期的早期阶段开始增加,在妊娠晚期达到最高,具体可参见《WHO Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology》。围产期毒性试验结果应在上市申请时提交。在适当的情况下,还应在III期临床试验前完成对雄性和雌性生育力潜在影响的评估。
3.5 非临床评价中的其它考虑因素
ADE:ADE的潜力应主要通过体外机制研究进行评估,无需专门进行动物研究,但如果存在该疾病的动物模型,则可以将ADE作为PD概念验证研究的一部分进行评估。
杂质:终产品中的杂质可能会引起安全性问题。这些杂质可能与产品相关且特性与目标产品不同(如mAb分子变异体、聚集体或片段),或者可能与工艺相关(如培养基成分或宿主细胞蛋白)。无论来自细菌、酵母、昆虫、植物或哺乳动物细胞的宿主细胞污染物,都存在潜在的风险。建议最好依靠质量控制和生产工艺来最大程度地减少杂质的存在,而不是通过非临床研究来评估其潜在影响。
生态毒性/环境影响:通常认为mAb对环境没有特别的危害,预计会通过分解代谢途径完全代谢,肾脏排泄可忽略不计。但对于某些化学修饰或偶联的单克隆抗体应进行全面的环境风险评估,除非另有说明。
过敏反应:尽管在人体中并不常见,但静脉注射mAb等蛋白质类产品可导致各种从轻度到重度不等的超敏反应,其分子机制可能不同,且大多未知。在动物研究中也可能观察到类似的超敏反应和输液反应,但这些反应可能无法反映此类反应在人体中发生的风险。豚鼠过敏性试验结果对蛋白质产品一般呈阳性,但通常无法预测人体的反应,因此不建议开展此类试验。
免疫毒性:通常不需开展,但如果在PD或毒性研究期间观察到mAb对免疫系统产生任何不良影响,并且这些不良影响可能导致宿主对病原体的抵抗力下降,则应考虑进行免疫毒性研究。
4总结与展望
虽然上市的单克隆抗体产品很多,但目前仅有少数获批用于治疗或预防感染性疾病。自2019年新冠疫情暴发以来,国内外批准用于预防和治疗感染性疾病的单抗药物数量逐渐增加。用于感染性疾病与非感染性疾病的单抗类药物在拟定适应证、药物靶抗原等方面均不同,故非临床评价亦存在差异。WHO于2023年发布了此指南,与常规用于非感染性疾病如肿瘤或自身免疫疾病的单抗类药物相比,该指南对拟用于感染性疾病的单抗类药物在非临床评价方面提出了新的或不同的关注点,特别针对动物种属选择、药效学、耐药性、ADE、非预期交叉反应性、生殖毒性等研究与评价予以了充分的阐述,同时对在公共卫生紧急情况下的简化非临床研究提供了一些特殊考虑。
本文针对该指南对用于预防和治疗感染性疾病的单抗类药物应当如何开展非临床评价进行了详细的介绍,以期为我国此类新药的非临床研究和评价提供参考。在药物研发过程中,鼓励申请人就试验策略、设计、结果分析等与审评机构沟通交流。希望未来在工业界和监管机构的共同推动作用下,能够提高该类药物的研发水平,促进产业化进程。

来源:Internet


