您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-07-24 20:08
摘要
为了提高患者依从性,皮下注射剂型是治疗用重组蛋白类药物差异化开发的重要方向之一。近年来,其临床试验申请和上市申请数量显著增加。而皮下注射剂型产品因治疗用重组蛋白类药物本身的固有属性、高浓度制剂对产品质量和稳定性的潜在影响、以透明质酸酶为代表的特殊辅料的应用等因素,在制剂处方开发、辅料质量控制、制剂质量控制和稳定性研究等方面与静脉给药剂型的药学研究相比存在一些特殊考量。本文汇总了2020年以来皮下注射剂型治疗用重组蛋白类药物申报情况,对药学专业审评中的特殊考量展开讨论,并提出该类药物研发与生产过程中可能存在的挑战,以期通过研发生产机构和监管机构共同努力,强化技术交流,为患者提供安全有效、质量可控、使用便捷的新型治疗用重组蛋白类药物。
关键词
皮下注射; 治疗用重组蛋白类药物; 制剂处方; 透明质酸酶
正文
由于治疗用重组蛋白类药物(therapeutic recombinant protein drug,TRPD)研发的不断深入和市场竞争的日益激烈,差异化设计逐渐成为药品开发的新方向。既往TRPD的主要给药方式为静脉注射或静脉输注(intravenous,IV),这需要洁净的给药环境、受过专业培训的人员和较长的时间成本[1]。相较于此,皮下注射(subcutaneous injection,SI)剂型不但在便捷性和依从性上具有显著优势,而且对血管堵塞或心血管脆弱的患者更为友好。以代表性的产品曲妥珠单抗为例,以固定剂量给药的SI剂型在安全性、有效性方面不劣于静脉给药制剂,而在患者依从性上,PrefHER研究显示91.53%的患者由于节约时间、减少疼痛等各方面原因,倾向于采用SI剂型; 而倾向于采用静脉给药方式的患者仅占6.78%[2]。因此,TRPD产品的SI剂型开发已成为新的研究重点。据报道,2008—2017年间,美国FDA批准的单克隆抗体产品中约36%为通过皮下注射给药,至2017年这一比例已经超过了60%[3]。
差异化的研究方向提供了新的机遇,但由于TRPD本身的固有属性,其SI制剂的开发仍存在挑战,例如: 相对分子量较大,已经超出毛细血管的吸收能力,而细胞外基质的复杂结构和组成又阻碍了其通过其他方式进行吸收[4]。因此,为了提高生物利用度,研究人员不得不通过各种形式的制剂处方调整或对活性物质本身进行改造以改善其成药性,而这些变化正是其药学研究的特殊之处。本文将结合目前已在国内申报上市的SI剂型TRPD案例,从药学专业角度出发,提出一些审评引发的思考。
1国内申报情况
根据公开数据查询可知,2020年以后在国内申报上市的SI剂型TRPD共计13个受理号(见表1),涉及10个品种,其中托珠单抗、达雷妥尤单抗、恩沃利单抗和曲妥珠单抗的SI剂型已获批上市,其余品种均仍在审评中或已终止审评程序,目标靶点则主要集中于肿瘤和免疫治疗领域。
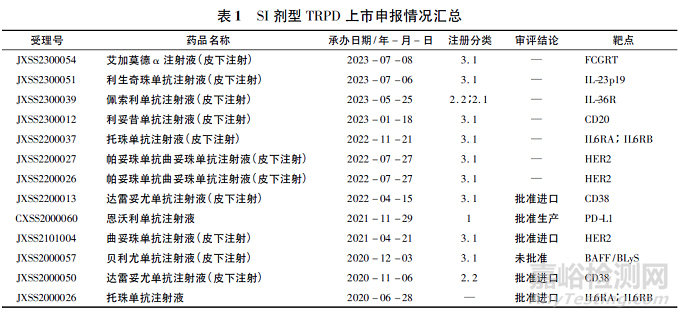
从审评实践上,目前在国内获批上市的SI剂型TRPD均已在国外获准上市,在积累了相对充分的生产和质量控制经验、具备了一定的上市后评价数据后来我国进行申报,因此申报资料总体较为完善,药学专业的研究也相对充分,而国内企业在该领域申报数量则相对较少。近几年,伴随审评审批制度改革和对于创新药物的大力支持,这一趋势正在发生改变。跨国制药企业的申报策略逐渐转变为国际多区域同期申报,国内企业的临床试验申报数量也在逐年递增。
2药学专业审评思考
SI剂型TRPD的药学质量控制要点与常见的静脉给药制剂相比较不存在本质区别,但针对该类产品的特点,需要重点关注以下研究内容。
2.1 制剂处方开发
目前已申报的SI剂型TRPD大多以已上市的IV剂型为基础开发而来,两者在制剂处方组成方面存在显著差异。其中,目标蛋白含量远高于静脉给药剂型是SI剂型制剂处方最显著的特点,后者的活性蛋白含量往往能达到100~200mg·mL-1,系同类静脉给药制剂的6倍以上。因此,SI剂型制剂处方开发的目标是在确保高浓度制剂稳定性的前提下,避免其在黏度、递送过程甚至免疫原性方面产生负面影响。
SI剂型制剂处方开发思路与静脉注射剂型制剂基本一致,一般采用阶梯式研发或多因素正交分析,对制剂的pH、缓冲盐体系、稳定剂及促吸收剂逐一或分组进行筛选。具体研究方案一般基于不同贮存条件,如各项强制降解或影响因素研究中,样品的纯度、电荷异构体、不溶性微粒等关键质量属性的变化情况确定最终的制剂处方。其特殊之处在于,为了维持高浓度制剂中目的蛋白的稳定性,近一半的产品会在制剂处方中增加一定量的非还原糖,如蔗糖等[5],而单克隆抗体等重组蛋白产品长时间与蔗糖接触可能会导致其糖化水平发生变化,即使是冻干制剂也具有潜在的风险,并进而影响其电荷异质性、生物学活性甚至是免疫原性。此外,该类产品中常使用聚山梨酯80或聚山梨酯20以防止高浓度的蛋白质在不同物质界面或振摇过程中形成聚集,但潜在的高宿主蛋白残留水平也可能导致其氧化和水解降解过程的加快,并进而导致脂肪颗粒或可见异物的形成,需要在制剂处方开发过程中予以充分关注。对于给药体积较大的SI制剂,往往采用透明质酸酶作为促吸收剂,以增加其生物利用度,在实际生产中,可以以活性为标准投料或以质量为标准投料,但应严格控制其比活性,以确保处方的一致性。
2.2 辅料质量控制
总体上,SI剂型TRPD产品所使用辅料如已被《中华人民共和国药典》收载,其质量标准应不低于药典要求。在特殊辅料中,透明质酸酶作为影响该类产品药动学行为和药效学的关键要素,对于产品安全、有效、质量均一的影响程度不亚于活性物质本身,因此建议参照原液/原料药的要求对其进行质量控制。
目前,可以采用多种方式制备透明质酸酶,包括组织提取、原核表达或真核表达等。对于药物/药品的研发和生产,质量均一性是关注的重点,考虑到组织提取来源的透明质酸酶可能在杂质组成、免疫原性、生物学活性等方面存在不均一性,不建议采用该种来源的透明质酸酶作为TRPD产品的辅料,而采用原核或真核细胞表达的产品在质量均一度的控制上则具有明显优势。对于采用真核细胞表达的透明质酸酶,应特别关注其病毒污染风险,申请人应在供应商审计中重点关注其生产用原材料(含细胞库)的检定、关键工艺中间体的检定及病毒去除灭活工艺验证等方面,确保其不具有病毒污染风险,并合理拟定其入厂检定标准,至少定期对病毒性外源因子进行抽检。
质量研究方面,无论是自行生产还是外购透明质酸酶,其要求是一致的。检定项目上,建议包括蛋白质含量、生物学活性、比活性、纯度(建议至少包括RP-HPLC和SEC-HPLC检测)、电荷异构体组成及活性、有关物质、杂质(含产品和工艺相关杂质)、安全性因素(生物负荷、细菌内毒素、支原体及外源病毒)检定。翻译后修饰是透明质酸酶需要特别关注的问题,一方面是关键位点的氧化、酰化等; 另一方面则是N-糖基化修饰,文献报道经PNGase处理的透明质酸酶,其生物学活性下降幅度可达80%[6]。因此,对于具有翻译后修饰的透明质酸酶应开展更为全面的质量研究和生产工艺控制。
2.3 制剂质量控制和稳定性研究
总体上,SI剂型TRPD产品的质量标准应满足《中华人民共和国药典》相关要求,在此基础上,结合拟申报产品在关键临床试验阶段各项质量属性波动情况,合理拟定质量标准限度。例如: 渗透压检定中,静脉给药制剂约在50~350 mOsmol·kg-1不等,而SI剂型则基本集中在300~400 mOsmol·kg-1; 细菌内毒素检定中,静脉给药制剂一般限度为不高于0.21EU·mg-1,而SI制剂中,由于蛋白浓度较高且剂量单位不同,这一数值可能提高数十倍。
对于采用了透明质酸酶的产品,一般要求在制剂质量标准中增加透明质酸酶活性检定,目前主要SI剂型TRPD产品的检定方法大多基于透明质酸酶与酸化血清结合时形成的不溶沉淀物,并由此测定其浊度。以标准品稀释液的浊度结果生成标准曲线,进而计算样品中透明质酸酶的活性,该检项质量标准的主要制定依据即关键临床试验批次样品的检定结果。
针对SI制剂的处方特点,还有一些特殊关注点。例如: 在含有高浓度还原糖的制剂中,建议考虑制剂在长期储存条件下糖化水平的变化情况以及其对于本品安全性和有效性的潜在影响; 针对采用了聚山梨酯80或聚山梨酯20的制剂,基于其宿主蛋白残留水平,建议持续关注其可见异物和不溶性微粒的检定结果,并增加采用微流数字成像[微流成像(micro-flow imaging,MDI)或微流数字成像(micro-flow digital imaging,MFI)]方法的检定。此外,高浓度蛋白制剂在长期保存的情况下,还可能导致其他关键质量属性的变化,例如: Fc单链总氧化或重链Asn330脱酰胺修饰水平等,建议在早期的稳定性研究中对以上因素进行充分观测,并在上市申报阶段合理拟定稳定性研究检项和标准。
2.4 生产工艺过程控制和给药装置研究
SI剂型TRPD作为典型的高浓度制剂,黏度是其关键的质量属性之一,且在一定范围内药品浓度和黏度之间存在着指数关系。生产过程中,高黏度会给产品的制造过程如搅拌、过滤、灌装甚至是清洁等方面带来一定困难,因此其工艺验证更加关注制剂生产过程中产品质量均一性的控制,以及灌装过程无菌验证对可能发生的堵塞等意外情况所开展评估的充分性。在给药装置的功能性方面,制剂黏度可能会对临床使用过程中实际给药量产生影响,为了克服上述负面影响,可能需要在制剂处方开发过程中加以关注或在给药装置上进行创新。例如: 采用电动自动注射笔进行给药,此时产品的药学研究就需要将装置功能性纳入考量。如涉及多次给药装置,还应具有医疗器械注册证,或在上市申报阶段提供相应的器械研究资料,以供药械联审。
2.5 生物类似药研发
目前,多个SI剂型的TRPD产品已成为生物类似药开发的重点方向,如达雷妥尤单抗等。该类产品在申报过程中常见问题有2类,一类是某申请人已经开发了该类产品的静脉给药制剂,基于其原液生产工艺未发生变更,且拟申报产品和已获批开展临床试验或上市产品在物质基础上的一致性,希望免于开展拟申报产品和原研参照药的全部或部分质量属性相似性研究,包括特征鉴定、杂质研究和批放行检定对比研究。考虑到高浓度制剂及处方组成可能对产品的翻译后修饰产生不确定的影响,因此建议申请人采用拟申报产品的原研参照药,即SI剂型的原研参照药规范开展质量属性相似性研究,并根据产品特性,合理设置稳定性相似性研究项目; 另一类是申请人自主研发了关键辅料透明质酸酶,且在制剂处方中与原研参照药的用量不同。根据生物类似药研发与生产相关指导原则,拟申报产品应开展处方筛选研究,并尽可能与参照药一致,对不一致的,应有充足的理由。因此,对于透明质酸酶这类与产品安全性、有效性密切相关的关键辅料,可以基于产品自身的特性和充分的研究,确定其用量,并在申报时提供充分的研究数据以支持制剂处方的拟定。同时,建议采用一定批次的拟申报产品和原研参照药,进行相同的预处理后对其中的透明质酸酶进行对比研究。
3结语
TRPD产品的SI剂型开发是简化给药方式、提高患者依从性的重大进步。尽管这一技术的基础研究已经长达近20年,但目前该类产品的药学研究,特别是针对重要辅料透明质酸酶及最终处方制剂的关键质量属性的认识仍在不断深入,产品上市后的持续研究和上市后监管是加深业界和监管机构对该类产品认知的重要途径。希望申请人和监管机构共同努力,强化技术交流,为患者提供安全有效、质量可控、使用便捷的新型TRPD。

来源:Internet


