药物研发中杂质的研究贯穿其整个阶段,是保证药品质量的重要内容。药品中的杂质一般分为三类:有机杂质、无机杂质及残留溶剂。
有机杂质是指在药品的生产与储存过程中引入的有机化合物杂质,这些杂质可以是已知的、未知的、挥发性的或不挥发性的杂质。其来源多种多样,主要包括:降解产物、聚合物、原料药与辅料或内包材的反应产物、以及原料药制备过程中引入的起始原料、副产物、中间体、反应试剂、配位体与催化剂。
无机杂质是指在药品的生产过程中引入的无机物质,这些杂质通常是已知的,主要包括:反应试剂、配位体与催化剂、重金属或其它残留的金属、无机盐、过滤助剂、活性炭等其它物质。
药品中的残留溶剂系指原料药或辅料的生产中,以及制剂制备过程中使用的,但在工艺操作过程中未能完全去除的有机溶剂,一般具有已知的毒性。
杂质研究及控制是药品安全保证的关键要素之一,是药品研发中风险控制意识的重要体现,药品临床使用中的不良反应除了与药品本身的药理活性有关外,有时还与药品中的杂质有关,须严格控制。
ICH Q3药物杂质指导原则
药物原料或制剂中的杂质可能引起临床不良反应。杂质毒理学评估是药物研究的重要内容。化学合成药的原药和制剂中杂质的毒性,尤其是遗传毒性,是影响药物安全性的重要因素。
ICH (International Council on Harmonization of Technical Requirements for Pharmaceuticals for Human Use, 国际人用药品注册技术要求协调会)制定了如何控制药品杂质的指导原则,现行指导原则包括:
Q3A (R2):新原料药中的杂质(Impurities in New Drug Substances)
Q3A主要阐述了原料药中对杂质控制的整体思路、方法和限度。Q3A中规定了新活性药物成分(active pharmaceutical ingredient, api)中杂质需要进行报告、鉴定和界定的限度。如杂质含量超过鉴定限度或达到质控限度,药物研发者有义务对杂质的安全性进行评价。
Q3B (R2):新药制剂中的杂质(Impurities in New Drug Products)
Q3B阐述了新药制剂中杂质控制,包括药物原料的降解产物或者药物与赋形剂和/或直接接触容器的反应产物。Q3B中规定了新药制剂中降解产物的报告、鉴定和界定的限度。
Q3C (R8):杂质:残留溶剂的指导原则(Impurities:Guideline for Residual Solvents)
Q3C引入了每日最大允许量(permitted daily exposure, pde)的概念,指药物中的杂质每日最大可接受而不产生毒性的摄入量,同时描述了PDE的计算方式。Q3C依据可能的健康风险程度,将常见的药物制剂中残留溶剂分为3类,并规定了相应残留溶剂的PDE值,如二甲苯的PDE值为21.7 mg/天。
Q3D (R2):元素杂质指导原则(Guideline for Elemental Impurities)
Q3D对元素杂质的控制思路和策略类似于残留溶剂的控制。根据元素杂质的毒性和制剂中存在的可能性,Q3D将24种元素杂质分为四类,并给出不同暴露途径下相应PDE值。
毒理学风险评估
传统的毒理学评估主要是基于动物试验,但体内试验复杂且费用高昂,同时由于保障动物福利等原因,故在化学品安全评估中,需要优先考虑减少动物测试,要求更积极地采用动物替代方法来获取化合物特性数据。化合物的毒理学风险评估是利用相关数据库和文献资料,针对特定化合物进行风险评估的方法。其检索评估的成本远低于生物学试验。因此结合文献资料和数据库进行毒理学风险评估已逐步代替生物学试验。部分监管单位如美国FDA也建议结合化合物的毒理学数据来开展风险评估。
1.毒理学评估流程
毒理学数据来源:
权威机构文件:ICH、FDA、U.S.EPA、WHO/JECFA、EFSA、OECD和ATSDR等
公开数据库:ECHA、HSDB等
2.非基因毒杂质的评估(PDE法)
对于非基因毒性化合物或具有实际阈值的基因毒物质,根据ICH Q3C和ICH Q3D指导原则推导PDE值。
PDE值是由POD(Point of Departure)结合不确定因子推导得到,PDE的计算公式如下:
PDE = POD × BW ÷ (F1 × F2 × F3 × F4 × F5 ×F6)
POD是检索获得的人类或动物相关毒理学试验的数据,是在对化合物的全身毒性、基因毒性、致癌性、生殖发育毒性、神经毒性等生物学终点进行全面考察,并依据产品的接触途径、接触人群、接触时间等信息来综合判断选定的,如NOAEL (No observed adverse effect level),LOAEL (Lowest observed adverse effect level),BMDL (Benchmark dose low),RfD (Reference Dose)等。
BW为质量调整系数,ICH假设任何性别的成人体重均为50 kg。
F1~F6:不确定因子,对选择的POD进行种内差异、种间差异、暴露时间、途径转换等修正。
3.基因毒杂质的评估
基因毒性杂质(genotoxic impurity,GTI)是指药物中能直接或间接导致DNA受损引起基因突变,并具有致癌性或者潜在致癌可能性的一类杂质。基因毒性杂质是受到药品监管机构和制药企业重点关注和控制的对象,由于其较一般杂质具有微量水平就存在潜在致突变性和致癌性风险的特点,需要严格控制其在药物中的含量以保证药物质量与临床应用的安全性。
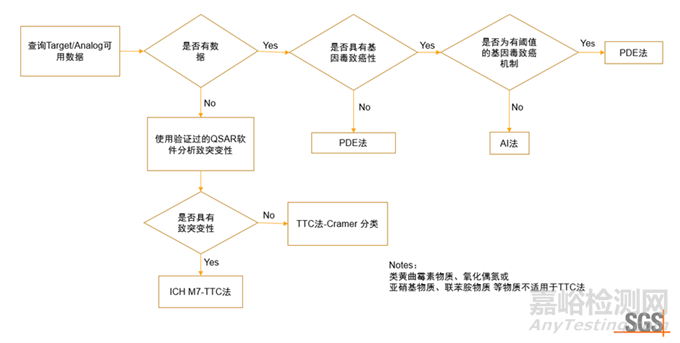
药物杂质研究中,首先基于警示结构,鉴别并分离出其中可能的基因毒性杂质。通过数据库文献检索、定量结构-效应关系(Quantitative Structure-Activity Relationship, QSAR)评估和体内外相关毒理学试验,对可能存在的基因毒性杂质进行分类并确定可接受的限度标准。
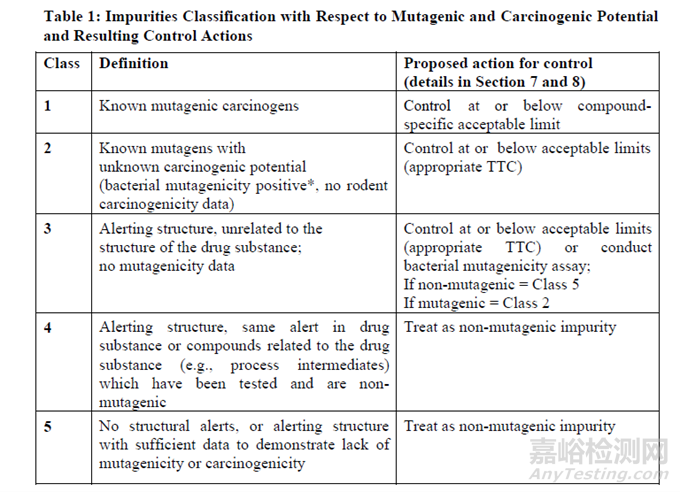
ICH M7 (R2) 评估和控制药物中DNA反应性(致突变)杂质以限值潜在致癌风险(Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk) 根据杂质的致突变性和致癌性分为5类。
3.1.有数据物质的限值推导(AI法)
对于已知的基因毒性致癌类化合物,可以根据其致癌性数据(TD50或SF)和线性外推的方法计算出该化合物特定的AI(acceptable intake)值。具体有以下两种方法。
TD50法:当动物致癌试验的数据可用时,将TD50(肿瘤发生率为50%时对应的剂量,相当于致癌风险水平为1:2)线性外推至致癌风险为10-5时对应的剂量,以此得到AI值。
AI(mg/kg/天)= TD50(mg/kg/天)÷ 50,000
SF法:斜率因子(slope factor)是暴露于1 mg/kg/天致癌化合物的单位癌症发生率。当斜率因子可用时,可以通过计算终生致癌风险为10-5时对应的剂量得到AI值。
AI(mg/kg/天)= 10-5 ÷ SF(mg/kg/天)-1
对于有数据支持为非基因毒性化合物或具有实际阈值的基因毒物质,根据ICH Q3C和ICH Q3D指导原则推导PDE值。计算方法同上述非基因毒物质PDE推导方法。使用该方法时需要更多的毒理学数据,证明阈值机制的存在。
3.2.无数据物质的限值确定(QSAR软件预测和TTC法)
根据ICH M7中的描述,危害性评估首先通过对数据库和文献的检索获得杂质致癌性和细菌致突变数据对实际和潜在杂质进行初步分析,并将其归为1、2或5类。如果无法获得化合物的致突变和致癌性数据及分类依据,则可进行定量结构-效应关系(QSAR)预测,如预测细菌回复突变性。常见的QSAR模型可分为专家知识模型(rule-based model)和统计学模型(statistic-based model)。专家知识模型是基于专家经验制定的各种规则所建立的;而统计学模型则是基于回归分析、偏最小二乘、神经网络等数学模型所建立。如果经两种互补的QSAR方法(专家知识模型和统计学模型)预测均无致突变性,则可认为该杂质没有致突变性,不建议做进一步的检测,可以判定该杂质为5类。
当缺乏足够的毒理学数据推导化合物的AI值,且QSAR预测细菌回复突变为阳性时,应使用M7 TTC法。ICH M7中的毒理学关注阈值(Threshold of Toxicological Concern,TTC),低于该水平时,可以认为暴露于该化合物对人类健康无明显风险。在使用TTC评估原料药和制剂中致突变杂质的可接受限度时,终生限度为1.5 μg/天。若存在同类基因毒杂质,可根据多个杂质的可接受摄入量来制定限度。
对于短于终生给药的药品中致突变杂质的TTC值可调整为更高剂量,不同给药时长的单个杂质TTC如下:
|
治疗期 |
≤1月 |
>1月-12月 |
>1-10年 |
>10年 |
|
TTC(μg/天) |
120 |
20 |
10 |
1.5 |
3.3. 遗传毒性试验
当QSAR软件评估致突变得到阳性结果时,也可进一步进行体外评估(如:Ames试验)。如果Ames试验为阴性,则该杂质应归为5类。如果Ames试验结果为阳性,后续应进行体内试验,明确体内致突变风险。
小结:
PDE法多用于非基因毒致癌物。对于致癌性数据为阳性的致突变杂质,需使用AI法进行评估。在目标化合物数据无法获得的情况下,可以通过QSAR预测并使用TTC值进行评估。
毒理学风险评估的应用
1.药包材相容性
药物在储存过程中,有效成分会持续与主要包装系统的材料发生相互作用。包装材料中的物质可能会迁移到药物中,对药效产生不利影响或者导致毒性。可提取物和可浸出物(E&Ls, extractable and leachables)研究可提供其相互作用的数据,从而评估其潜在风险。任何测得超过分析评估阈值(AET)的化合物都应该被识别出来并报告其检出量,该检出量应根据药品使用方式转化为患者潜在暴露的估计值,再与化合物对应的允许限量(如PDE)进行比较,判断风险。
2.杂质限度
当企业在新药研发过程中,某特定杂质超出了ICH Q3的限度要求,应当除去这个杂质或降低到ICH Q3要求的限度水平,但这可能是非常困难或代价非常高的。那么可以根据该杂质的毒性水平制定其PDE值。PDE与限度本质上等同,都用于表示杂质控制的水平,不同在于ICH Q3中的限度通常以百分比(%)表示,而PDE为某种化合物的特定限度,是根据其毒性数据专门推导得到,通常用μg/天表示。PDE除以产品的最大日服用剂量,得出特定化合物的安全限度值。
3.共线生产中的残留限度
当一个共用设施替代多个专用设施生产不同药品时,首先要解决如何防止交叉污染的问题。产线上的一种药品的残留对同产线其他药品使用的患者来说可能属于污染物,从而带来潜在风险。因此,在共线生产过程中建立科学的清洁限度尤其重要。基于健康的暴露限度(health-based exposure limit,HBEL,如PDE值)常被用于共线生产中用于设定清洁验证的残留限度。
传统方法:1/1000最低日治疗剂量、10ppm方法等。统一限度,但存在风险和局限。
HBEL法:使用PDE、ADE(acceptable daily exposure)等,更加科学合理。
2014年欧洲药品管理局(EMA)发布的共用设施中生产不同药品的暴露限度设定指南《Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities》和2020年世界卫生组织(WHO)发布的清洁验证指南草案《Points to consider when including Health-Based Exposure Limits (HBEL) in cleaning validation》,均提出需要使用以HBEL为基础的残留限度。
2023年,中国国家药品监督管理局药品审核查验中心(CFDI)发布《药品共线生产质量风险管理指南》,其中提到“活性物质残留限度标准应当基于产品毒理试验数据或毒理学文献资料结合实际生产情况建立。相对于传统方法设定的限度来说,基于健康的暴露限度(HBEL)的可接受标准(如PDE值)在评估清洁残留数据时更具有科学性和优势”,体现出国内对于HBEL方法的关注度也逐渐提高。