您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2021-10-11 23:22
内容提要:通过查看检验报告中载明的标准信息、检验数据、适用项目,比对出厂检验报告,发现出厂检验项目和检验设备的缺陷,介绍检验报告在医疗器械现场检查时对质量控制部分的检查指引作用。以检验报告为线索和依据,可以帮助医疗器械检查员在现场检查中快速、准确的识别质量控制部分的缺陷。
关 键 词:检验报告 医疗器械 现场检查 质量控制
质量控制作为《医疗器械生产质量管理规范》(以下简称“规范”)的重点章节,规定企业应当建立质量控制程序,规定产品检验部门、人员、操作等要求,并规定检验仪器和设备的使用、校准等要求,以及产品放行的程序[1]。该章中共包含6条规定,其中有4条均涉及检验,由此可见,检验是质量控制的重要组成部分。原国家食品药品监督管理总局于2012年4月18日发布了《关于印发全国食品药品监管中长期人才发展规划(2011—2020年)的通知》[2],文中提及对专职检查员队伍的建设要求,目前的检查员队伍结构主要以长期从事监管和审评认证工作的人员为主,具有丰富的现场检查经验,但较为缺乏检验相关知识,而建设专业检验检测队伍同样重要,在医疗器械高速发展的当下,检验检测工作量也逐年大幅增涨,根据职能划分,现场检查工作中引入大量检验人才在短期之内无疑是不现实的,而且检验技术人员在现场检查中还需扩充较多体系相关知识,要快速进入检查员角色也需要时间。本文将介绍如何通过查看检验报告发现现场检查中有关质量控制部分的关键问题,帮助检查员在抛开专业的情况下,仍可以顺利完成对检验有关内容的检查工作。
1.通过查看检验报告发现产品出厂检验的缺陷
“规范”第五十八条规定,企业应当根据强制性标准以及经注册或者备案的产品技术要求制定产品的检验规程,并出具相应的检验报告或者证书[1]。据此,检查员需熟悉产品适用的强制性标准或经批准的技术要求,但由于医疗器械标准较多、内容复杂,尤其是有源医疗器械的常用安全标准,需要有多年工作经验的检验人员才能理解透彻,检查员在查看出厂检验报告时往往会感觉困难,而出厂检验是现场检查的必查项,亦是成品放行的重要关注项,若此部分出现缺陷,将直接影响产品质量,甚至造成上市后监督抽验不合格。由于检验的相对专业性,企业在检验标准的理解上也存在不足,从全国各大医疗器械检验机构的注册检验情况了解到,送检样品一次性合格率较低,送检采标完整的技术要求更为少见,企业通常依赖检验机构对其技术要求预评价的同时作修订。因此,企业的出厂检验规程时常会出现与强标或技术要求不一致的情况。现通过以下示例详细介绍。
1.1案例一
某数字式移动X射线设备生产企业,在生产许可检查中,通过查看注册检验报告电介质强度内容核对企业出厂检验规程和出厂检验报告,发现规程和出厂报告中显示的电介质强度要求为3000V,而注册检验报告要求为4344V,出厂检验规程要求明显低于技术要求中所采电气安全标准GB 9706.1-2007的试验电压值[3]。经询问得知,企业错误理解标准,且未关注取得的合格注册检验报告内容,故仍沿用错误的出厂检验规程。考虑到仍在申请许可阶段,未造成后果,故归为一般缺陷,若此次检查为上市后监督检查,此缺陷将进一步评估。注册检验报告内容见图1。
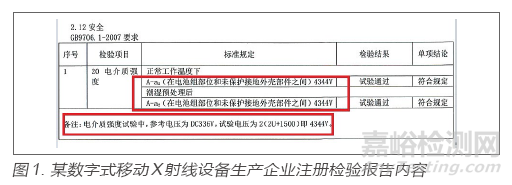
在检验报告中可看到理解起来较为晦涩的专业术语和计算公式,而检查员在没有足够的检验知识时,完全可以通过仔细比对注册检验报告和出厂检验规程和报告发现缺陷。
1.2案例二
某电动手术设备生产企业,在监督检查中,通过查看注册检验报告接地阻抗内容核对企业出厂检验规程和出厂检验报告,发现多份出厂报告中显示的接地阻抗实测值为0.03~0.05Ω,结论为符合要求,而注册检验报告显示该项为不适用。检查组因此怀疑出厂检验结果真实性,要求企业专职检验人员现场重现检验过程,而重现结果显示无法测出接地阻抗值,产品为全塑料绝缘外壳密封,无可触及接地金属部分,故无法测出接地阻抗属正常现象,该出厂检验项目数据不真实。检查组通过核对注册检验报告全面抽查了出厂检验项目,发现多个安全相关检验项目数据不真实,考虑到该产品用于人体开颅手术,并已上市销售,属于监督检查关键缺陷项。注册检验报告内容见图2。

在检验报告中“—”代表不适用,通常不会引起关注,出厂检验项目一般不会将不适用项列入考核项,而当发现出厂检验项目将不适用项判定为适用并出具实测数据结果时,需重点关注。
1.3案例三
某IVD设备生产企业,在监督检查中,该企业出厂检验报告中对电气安全项目GB 4793.1-2007[4]第6.3.1条“可触及零部件的允许限值正常条件下的值”的检验情况为“小于0.5mA有效值,小于0.7mA峰值”,并测量了电容值,查看注册检验报告,同样的项目显示电流限值不适用,电压限值符合要求。检查组在查阅国标后发现,标准要求只有当电压值超出规定值时,才测量电流值和电容值,该企业未按强制性国标规定的程序进行检验,但考虑到企业所测内容与未测内容均表示对同一项目的考核,对风险有一定程度管控,故考虑为一般缺陷项。检查员在发现同样的检验项目出现截然不同的要求和结果描述时,应查询此项目对应的国家标准,寻找不一致的原因。注册检验报告内容见图3。
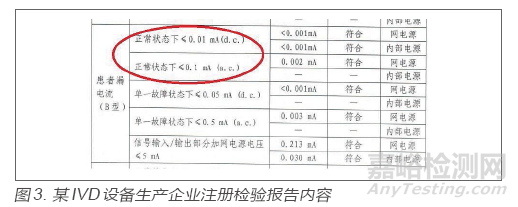
出厂检验报告未体现6.3.1a)的要求,仅有6.3.2b)的内容,而考核的项目一致,此时,建议检查员应查询国家标准对应项目,分析出现异常的原因,根据风险判断差异的属性,作出客观评价。
2.通过查看检验报告发现有关检验设备的缺陷
“规范”第二十一条规定,企业应当配备与产品检验要求相适应的检验仪器和设备,主要检验仪器和设备应当具有明确的操作规程[1]。此条规定在“规范”对设备要求的章节中,但通常在检查这一项时,需查看出厂检验规程、出厂检验报告和计量证书,这也是查质量控制部分的主要途径,因此两部分内容应同时检查[6]。通过查看注册检验报告对判断检验设备是否适应产品检验也是十分有效的,但检查员时常会忽略其关联性。现通过以下示例详细介绍检验报告与设备的关联问题。
2.1案例一
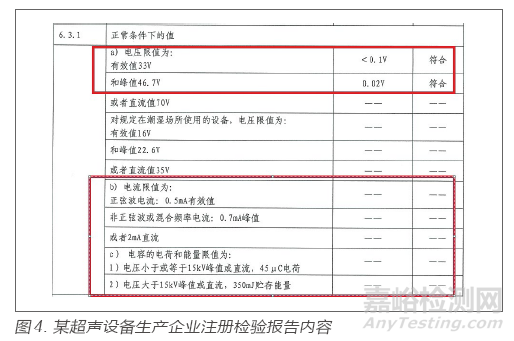
某超声设备生产企业,在注册体系核查中,查看注册检验报告,注册检验报告在患者漏电流的测试时体现了直流分量(d.c.)和交流分量(a.c.)的测试结果,而成品检验报告仅测试了交流分量,该设备的计量证书中也未体现直流部分的校准结果,由此引起了检查组对漏电流测试设备的关注,经核实设备实物和使用说明书发现企业采购的漏电流测试仪不具备直流分量测试功能,不能满足出厂检验需要。注册检验报告通常会对有限值要求的项目出具具体测试值,通过追溯测试值对应的检验设备,可核对设备是否具备出具相应测试值的功能。注册检验报告内容见图4。
案例二,某热敷贴类无源产品生产企业,在注册体系核查中,查看产品注册检验报告中对尺寸厚度的测试值,显示为“5.11mm”,出厂检验报告中对厚度的测试结果为“5.13mm”[5]。查该项目测试所需仪器游标卡尺实物和计量证书时,发现该游标卡尺测量精度为0.02mm,测量结果不可能出现单数尾数,出厂检验报告上填写的检测游标卡尺使用编号和型号与所查卡尺一致,从而得知企业随意填写检测结果,而注册检验报告出现的单数值是因为存在精度为0.01mm的游标卡尺,但企业并未采购。类似的情况经常会在检查中出现,如:无菌企业在测试液体pH时,采用普通pH测试试纸,精度为0.5,而通常会发现“7.2、8.1”之类读数。主要原因在于企业检验人员在千篇一律的重复检验工作中,通常会产生惰性,随意填写真值附近的相似值是普遍现象,但由于未考虑到设备精度和测量范围等实际情况,出现检验数据真实性问题。因此次检查为注册体系核查,并未正式上市,故要求企业整改后复查。该缺陷项不属于设备部分,但可通过重新配置精度更高的设备来整改。
3.小结
检验报告反映了产品在注册时的真实检验情况,尽管报告内容专业性较强,标准术语较多,检查员不可能面面俱到清楚所有技术知识,但合理的运用检验报告作为检查依据是有必要的。通过查看检验报告中的载明内容,结合产品技术要求、设备档案可以较容易发现出厂检验和设备方面的缺陷。需注意的是,检查员在发现缺陷时,应客观全面的查找原因,围绕现象搜集完整的证据链,对缺陷的判断应从局部放大到整个体系的风险评估,不应狭隘的根据“规范”条款和“规范”指导原则星号项草率出具关键缺陷结论。

来源:Internet


