您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-08-27 15:32
降解杂质的研究在整个药品研发过程中占着举足轻重的地位。自ICH M7颁布以来,直接将其推向了顶峰,成为行业研究的热点和难点。2018年华海药业NDMA事件依然在耳畔回响,时刻鞭策制药人砥砺前行。本文着重关注致突变杂质风险评估的深度而对风险评估的广度不作论述。
一、ICH M7关于API和制剂致突变降解杂质的政策解读
ICH M7指出:“制剂中实际的降解产物包括那些制剂在拟定的长期储存条件以及内包装和外包装储存期间观察到的高于ICHQ3B报告限度的杂质,还包括那些在制剂生产过程中产生的杂质。如果实际降解产物水平超过ICH Q3A/3B所述的鉴定限度,则应进行鉴定。有些低于鉴定限度的降解产物可能也要进行鉴定。”这段话清晰的告诉我们降解产物中若有痕量致突变杂质,需要进行评估和控制。然而低于鉴定限的痕量杂质如何进行鉴定、如何进行评估与控制,是当今世界药品研发一直在探讨的难点。
ICH M7还指出:“对于已经定性为具有致突变的潜在降解杂质,一定要了解该降解途径与原料药和制剂的生产工艺和/或其拟定的包装和存贮条件的相关性。”换言之,如果对于API和制剂中致突变杂质的研究深度不够,可能导致药品的包装和存贮条件不合适,直接结果就是药品的安全性无法得到保障。可想而知,若出现这样的情况,审评通过的可能性就极低,数十万的审评费就此挥手告别。
二、评估致突变降解杂质的一般思路
本文参考有关文献,探讨如何依据ICH M7的要求对API和制剂中致突变降解杂质进行评估。ICH M7明确指出:“相关降解途径的知识,例如来自于化学降解原理、相关的强制降解实验和研发期间的稳定性研究,可用来指导选择需要评估致突变性的潜在降解产物。”这是评估致突变降解杂质的核心指导思想。为了确定潜在降解产物的相关性,需要进行稳健的基于科学的风险评估试验研究1:
通过精心设计的强制降解试验研究来进一步检查;
加速稳定性试验;或精心设计的动力学等效更短期的稳定性研究;
通过原料药和制剂的长期稳定性研究确认。
由ICH Q1A所推荐的强制试验研究能够用来获取一组潜在降解产物,所产生的降解产物可以包括在风险评估中。这些强制降解产物在ICH长期和加速稳定研究中可能存在,也可能不存在,属于潜在降解产物。
评价ICH 加速稳定性试验结果(40℃/75%RH6个月)也可以揭示应该评估的降解产物。其研究包括一系列的稳定性条件:从敞口存贮到最终包装产品进行的相应研究。这些条件所形成的的降解产物应进行致突变杂质的风险评估,属于潜在降解产物。
在拟定的长期稳定性存贮条件(按照ICH Q1A和M7执行的一级和二级包装)所形成的降解产物也应进行致突变杂质的风险评估,属于实际降解产物。
在药物中致突变杂质是痕量存在,其发现具有极高难度。如果没有开发出针对性的检测方法,就无法发现这种比微量还要低几个数量级的杂质。有机化学降解原理可以为我们找到恒量降解杂质的可能性,这一方法的有效性取决于理论和经验的强弱。如果要想弥补短板,可以借助于专业软件帮助我们全面而深刻的理解特定的药物分子结构可能存在的降解产物。国外著名的化学降解预测软件Zeneth就是一款基于大量的化学反应数据库所提炼的的规则进行潜在降解产物预测的软件。软件预测的优点在于我们可以更为全面的理解原料药和制剂的降解路径:酸、碱、氧化自由基诱导剂、过氧化氢、重金属、光、溶液pH、辅料、杂质等等因素中哪一个或哪几个因素起了作用。这样就避免了人为经验和理论水平的不足导致评估的缺陷。当然缺点就是要评估的降解产物远多于实际存在的降解杂质。这显然增加了药品研发的成本。因此,随之而来的问题就是使降解杂质的研究对象更加聚焦且要尽可能的减少遗漏从而达到降低研发成本但不降低药品安全性的研发目的。基于此种需求,强制试验研究的新功能应运而生。
传统的强制试验(亦即强制降解)在制药工业中的功能有:获取关于原料药和制剂稳定性的知识和理解;识别降解路径;为制剂工艺的开发策略提供先验知识;开发指示稳定性的分析方法。ICH M7直接将强制降解试验研究的作用进一步拓展为:强制降解试验又可以提供降解杂质的研究对象。功能的变化迫使合成研究人员也需要掌握强制降解研究试验的规范。
三、评估致突变降解杂质的三种策略
强制降解的产物数目众多,ICH指导文件并没有就如何进行强制试验研究以及降解产物中哪些杂质为评估对象出台细则。不同的公司对确切的降解条件有不同的解读。Glaxo Smith Kline、Astra Zeneca、Lilly等制药公司的科学家总结了三种方法用以辨识降解路径(针对致突变杂质)和确立潜在致突变降解杂质的研究对象1。
方法一:观察加速和长期稳定性研究结果后进行结构鉴定。第一种策略简单易行,使用强制降解引导加速和长期稳定性研究。为强制降解研究而制备的样本用于开发指示稳定性的分析方法。强制降解会生成数量巨大的降解产物,降解产物的种类多和含量高使得降解物易于检测(使用已经开发的方法)。通过比较加速和长期稳定性的检测结果,依据ICH M7的指导原则确定哪些降解物需要结构鉴定。ICH M7指出:“如果实际降解产物水平超过ICH Q3A/3B所述的鉴定限度,则应进行鉴定。有些低于鉴定限度的降解产物可能也要进行鉴定”。在加速和长期稳定性研究试验中低于鉴定限的杂质也需要进行结构鉴定,这些杂质在强制降解试验中含量可能就会比较高,从而便于进行结构鉴定。这里要注意“有些低于鉴定限度的降解产物可能也要进行鉴定”是一个模糊的说辞,不利于指导研究工作的开展。
方法二:通过计算降解产物的相对含量进行结构鉴定。第二种策略使用强制降解研究来描述潜在降解产物和降解路径,对“主要”的降解产物进行鉴定。如何确定“主要”的降解产物则是第二种策略的核心。系统的方法是Alsante等人所做出的研究工作2。Alsante等人将“主要的降解产物”的定义为:一个降解物产生的量是总体降解量的10%,并且还要大于最大单个降解物含量的25%(图一和图二以实例说明)。这一策略已被Dow.L.K等人成功应用于开发实践,并且可靠地显示出主要降解路径,创建了合理数目的潜在降解产物(全面包含了实际降解产物)。表1进一步增加比较两种标准的细节。(关于降解程度的有关要求参见ICH Q1和相关文献。)
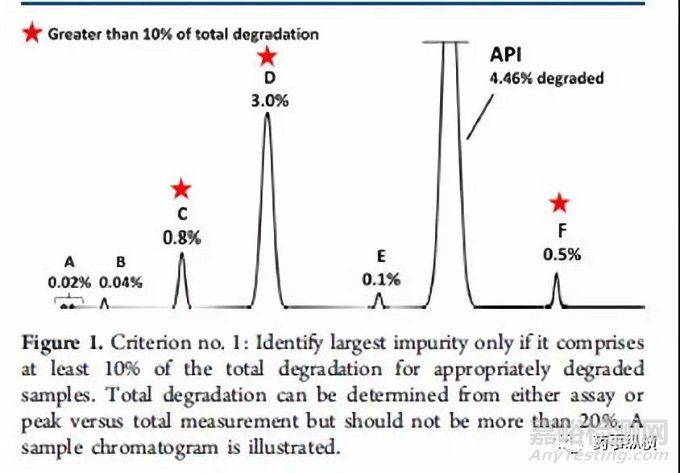
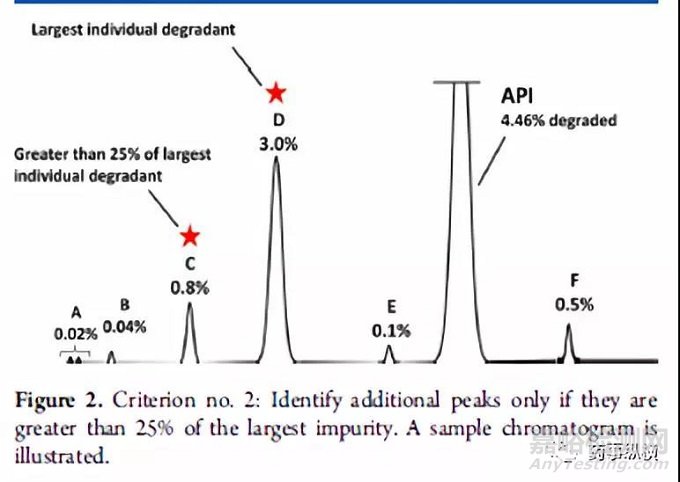
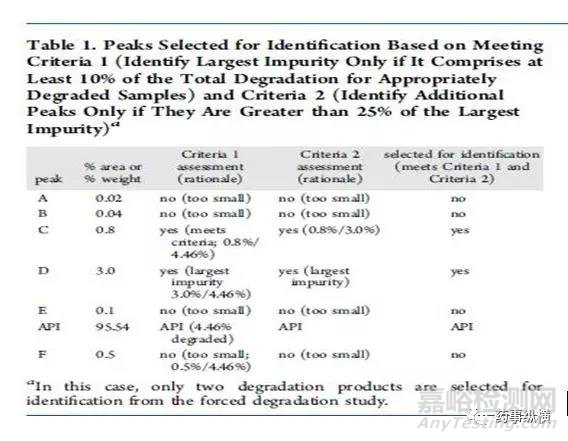
图片方法三:通过使用动力学等效和成比例放大的ICH Q3B限度进行结构鉴定。第三种策略是基于动力学等效的概念和ICH Q3B限度而开发的方法。这种策略要比上述两种更为复杂,但它可以减少需要观测和形成的潜在致突变杂质的数量,尤其是上述两种方法还没有完全建立起来的时候更为适用。感兴趣的读者请参考相关文献1,3,4。基于动力学等效实验得到的降解程度通常是5-10%,这个数据是货架期内可接受的降解程度(一般为2%)的2.5-5倍。由此获得动力学等效实验降解物的鉴定限为正常存贮条件下鉴定限的2.5-5倍。这种方法可以免去鉴定强制降解条件下产生的大量含量较低的降解物,而这些小含量的降解物与货架期内按照ICH条件存贮的情况下所产生的降解物没有任何相关性,从而减少了无用功。
以上三种方法各有千秋,相得益彰:第一种方法耗时较长且容易遗漏;第二种方法简单但工作量大(笔者在实际工作中第二种方法应用较多);第三种方法理论水平和实验设计能力要求较高,但目标性更强无用功更少。
限于笔者水平不正之处敬请指正。
参考文献:
1.MarkH. Kleinman ;Andrew Teasdale. Organic Process Research & Development. 2015,19,1447-1457.
2.Alsante,K.M.Adv. Drug Delivery Rev.2007,59,29-37.
3.Waterman,K.C.Int.J.Pharm.2005,293,101-125.
4.Waterman,K.C.Pharm.Res.2007,24,780-790.

来源:Internet


