您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-06-15 10:01
有关物质较含量测定而言是一个更加复杂和琐碎的体系,含量测定仅要求准确分析主成分,有关物质旨在研究所有应研究的杂质。
然而,研究没有那么容易,每个杂质有它的脾气 。
有关物质的研究思路
研究思路这个部分,应该由分析人员与合成人员共同讨论商定。项目开展前期,讨论会的频率会比较高,研究思路可能也会随着讨论而发生很多变化。
1.合成路线
API的工艺路线是杂质研究的关键,捋顺工艺路线,可以清楚每个杂质的来源和去向,更有针对性地给出杂质控制的策略。比如,哪些杂质可以仅在起始物料中分步被控制,后续API中无需再研究;比如,是否有些杂质在工艺中一直会有产生的可能性,那么仅在终产品中控制才能将其完全控制住,在前几步反应的研究中仅对其进行专属性的研究即可。
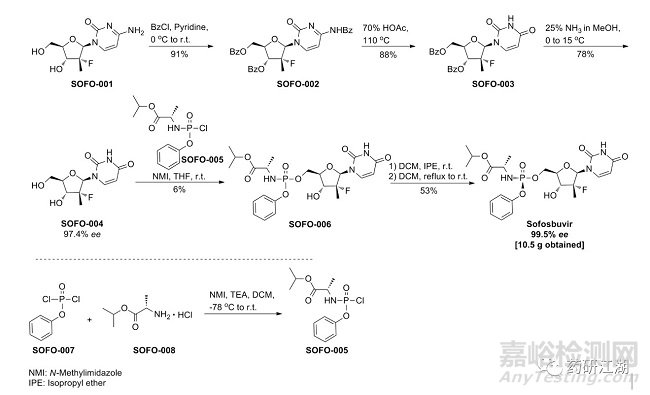
Sofosbuvir (索磷布韦)工艺路线
2.杂质谱分析
分析各工艺杂质是从哪一步得来的,如何产生;分析各降解杂质是由什么条件降解而来(要综合前期降解试验结果而考虑),列出杂质产生机理,然后再将杂质信息汇总成表格,推荐格式如下:
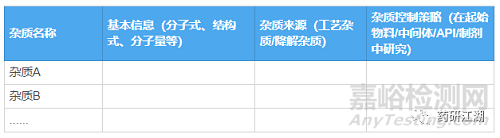
杂质限度的制定
ICH Q3A中关于原料药的杂质限度规定见下表:
|
最大日剂量 |
报告限度 |
鉴定限度 |
界定限度 |
|
≤2g |
0.05% |
0.10%或1.0mg(取最小值) |
0.15%或1.0mg(取最小值) |
|
>2g |
0.03% |
0.05% |
0.05% |
ICH Q3B中关于制剂的杂质限度规定见下表:
|
报告限度 |
最大日剂量 |
≤1g |
>1g |
||
|
限度 |
0.1% |
0.05% |
|||
|
鉴定限度 |
最大日剂量 |
<1mg |
1mg~10mg |
>10mg~2g |
>2g |
|
限度 |
1.0%或5μg (取最小值) |
0.5%或20μg (取最小值) |
0.2%或2mg (取最小值) |
0.10% |
|
|
界定限度 |
最大日剂量 |
<10mg |
10mg~100mg |
>100mg~2g |
>2g |
|
限度 |
1.0%或50μg (取最小值) |
0.5%或200μg (取最小值) |
0.2%或3mg (取最小值) |
0.15% |
|
根据参比制剂说明书或查找相关文献,可得知药品的最大日剂量,根据ICH指导原则的相关规定,对应查找杂质限度。值得说明的是,即使按照ICH指导原则来看,对应报告限度为0.05%,它只是针对检验这个产品是否合格、是否可放行时的报告限度,在研究阶段不建议将0.05%以下的小杂质忽略不计。
若真的忽略掉,很可能会忽略掉一些降解趋势或是产品的其他特性。
另外还要仔细对比和研究各已有标准规定的杂质限度,以目前审评趋势来看,各杂质限度推荐以最严格的限度来控制。
针对工艺杂质,放行标准和货架期标准一般来说是一致的。针对有明显降解趋势的降解杂质,可以将放行标准收严,以确保在稳定性过程中产品有关物质合格。
分析方法的确定
确定杂质的研究思路之后,可以开始技术性地进行方法开发。
1.已有标准和文献的参考与汇总
于仿制药而言,对已有标准的研究和分析是很重要的环节。在开发方法之前,应将已有标准和文献全面汇总分析,推荐列表如下:
|
模块 |
自拟标准 |
进口注册标准 |
USP |
BP/EP |
.. |
|
色谱柱 |
|
|
|
|
|
|
流动相 |
|
|
|
|
|
|
梯度洗脱程序 |
|
|
|
|
|
|
柱温 |
|
|
|
|
|
|
波长 |
|
|
|
|
|
|
流速 |
|
|
|
|
|
|
稀释溶剂 |
|
|
|
|
|
|
系统适用性 |
|
|
|
|
|
|
进样体积 |
|
|
|
|
|
|
供试品浓度 |
|
|
|
|
|
|
对照品/对照溶液 |
|
|
|
|
|
|
计算方法 |
|
|
|
|
|
|
限度要求 |
|
|
|
|
|
2.色谱条件的选择
首先,这个部分的研究,应基于对各已有标准中有关物质方法的了解。如果各标准中色谱条件完全不同,可分别进行重现。重现过程中主要关注:各杂质组分峰的分离情况、供试品检出杂质的灵敏度情况、峰型和基线的美观度、方法的省时省力程度,从中选择最优方法进行调整和优化。
但,有一点要注意的是,在重现已有标准的时候,一定要多想一想,这个色谱条件是不是已经研究透彻了,是不是真的做到了重现。比如有些进口注册标准,色谱柱的品牌和类型并未详细规定,实验者随意拿来一根色谱柱使用,峰型不好就将这个方法摒弃了,这是很武断的,一定要仔细对各项参数进行考察,也许不小心放掉的就是方法开发中的关键点。
有时候作为舶来品的国外标准或进口注册标准是会放些烟雾弹来迷惑当事人的,但是改变它的一些关键参数也会带来风险。比如无故提高供试品浓度,可能会带来峰型美观度的下降,杂质的检出过于灵敏,组分峰间分离度减小,而且这些风险和弊端可能会在日积月累的检验过程中日益凸显,后续再去修改和调整方法,又会浪费人力物力,得不偿失。因此,在调整方法时要考虑,是不是各方面都已考察完全,避免给自己后续带来不必要的麻烦。
在色谱条件优化时,应充分考虑杂质的理化性质和结构特点。比如:极性、酸碱性、溶解度、特征基团,这些特点会在调整色谱条件时给实验者很多启示。对色谱条件进行重现和优化之后,有关物质的分析方法就可初步拟定了。
3.检测波长的确认
选择检测波长,可使用PDA检测器的液相色谱仪,在进行方法确认时,直接提取各杂质定位溶液的紫外吸收光谱而定,当然也可以用紫外光谱仪分别扫描各组分对照品溶液(个人觉得比较麻烦,不推荐)。各杂质在所选波长处均应有较大吸收(有时综合考虑已有标准所规定的检测波长)。
4.溶液的配制方式
旨在考察提取的方法、次数、时间等因素。
当供试品稳定性不好或者主成分难以从制剂中提取出来时,应考察超声、振摇或其他方式,以及处理的时间长短。主成分提取的难易程度取决于主成分在溶剂中的溶解性和制剂工艺,应视情况而选择应有的方式。若配制原料药供试品溶液,一般仅考虑溶解性即可。
推荐每种方式平行考察2~3份样品,评估每种配制方式的杂质个数、单个杂质含量、杂质总量、主成分含量等,确保选定的配制方式可以将供试品提取完全,且提取过程中未发生明显降解。
提醒:最终选定的配制方式应有一定的耐用性,比如提取时间有小的变动或者换用超声仪、换用摇摆振荡器时不会影响样品的提取程度。还有一些情况下,在片剂或颗粒剂研细过程中,研钵也会吸附主成分,或是产生一些杂质,也许换用投片法可以规避这些问题。
5.溶液的处理方式
主要针对于制剂的供试品溶液,配制后进行离心/过滤处理。
推荐考察如下指标:
|
溶液类型 |
推荐考察处理方式 |
考察指标 |
|
|
空白溶剂 |
离心转速、离心时间、滤膜品牌、滤膜种类、不同滤膜批号等 |
离心与过滤后是否产生新的色谱峰,若产生,是否干扰各杂质和主成分峰 |
|
|
空白辅料溶液 |
同“空白溶剂” |
||
|
杂质混合溶液 |
离心与过滤前后各杂质峰面积的变化情况 |
||
|
供试品溶液 |
离心与过滤后杂质个数、单个杂质含量、杂质总量、主成分峰面积等变化情况 |
||
同时也要考虑具体操作的可行性。比如一些制剂的缓释材料会使过滤这个操作变得尤为困难,过滤一个供试品会让一个小姑娘累到怀疑人生,此时在离心不受影响的前提下,只能推荐采用离心方式进行处理。
6.总结和提示
上文写出的是一些比较常见的开发过程,当然每个品种有自己的特点,开发过程中要研究的可能会更多。比如有时在不同溶剂中,样品的稳定性会有所不同,那么溶剂的种类也要进行考察和选择。比如称样量和稀释体积是否也会对供试品产生影响。各位需全面关注分析方法的各项参数,甚至观察配制过程中的每一个现象,为方法开发的准确可靠性再添一把柴。毕竟人生已经够艰难了,能少挖给自己的坑,就少挖一点吧。
方法学预验证
把预验证这部分放在方法开发中作为一个标题来撰写,是想要提示各位,不要开发出一个方法后,就匆忙开展方法学验证,先进行一些预验证的试验也是非常有必要的。
1、强制降解实验可以帮助补充理论中没有被分析出来的降解杂质,避免后续稳定性过程中再产生,还要再重复研究一遍。
2、有些杂质,配制和处理方式会影响其回收率结果。
3、对于一些结构不稳定的物质,先期考察室温和冷藏放置的溶液稳定性,可避免验证过程中走不必要的弯路。
4、至于耐用性的预验证,更是不可忽视。

来源:铭研医药


