您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-02-21 21:12
经皮给药是一种重要的给药途径,皮肤递药制剂分为局部作用的传统制剂和现代经皮递药系统,前者包括软膏剂(ointments)、乳膏剂(creams)、糊剂(pastes)、凝胶剂(gel)、喷雾剂(spray)、涂剂(liniments)、气雾剂(aerosols)、巴布剂(cataplasms)、泡沫剂(foams)等。现代经皮给药系统一般指贴剂(patches)。
《中华人民共和国药典》2015年版四部贴剂通则中关于贴剂和透皮贴剂的描述如下:贴剂系指原料药物与适宜的材料制成的供黏贴在皮肤上的可产生全身或局部作用的一种薄片状制剂。其中用于完整皮肤表面能将药物输送透过皮肤进人血液循环系统起全身作用的贴剂称为透皮贴剂。
透皮贴剂具有许多优于常规注射剂和口服制剂的优点,例如,能够长时间持续释放药物,避免口服给药引起的血药浓度的峰谷现象,避免肝脏的首过效应和药物在胃肠道的降解,适用于睡着的、无反应的或者不能吞咽口服药物的患者(提高患者顺应性),发现不良反应时可随时中断给药。
本文就国内外上市透皮贴剂、欧美关于透皮贴剂仿制药的技术要求和实践等进行综述,探讨我国透皮贴剂存在的问题并提出建议,为我国透皮贴剂仿制药的研发和监管提供参考。
透皮贴剂概述
透皮递药系统(transdermal drug delivery systems,TDDS),又可称为透皮贴剂,起源于美国。20世纪70年代,全球著名释药技术公司美国ALZA公司开发了FDA批准的首个透皮贴剂东莨菪碱透皮贴剂(商品名:Transderm scop),1979年被FDA批准上市。
迄今FDA共批准了17种药物的透皮贴剂产品(包括单方和复方制剂,见表1),透皮贴剂用于治疗多种病症和疾病,包括由于绝经引起的中度至重度血管舒缩症状、阿尔茨海默病、注意力缺陷多动障碍、重度抑郁症、慢性疼痛的控制、恶心和呕吐的预防、预防心绞痛等。
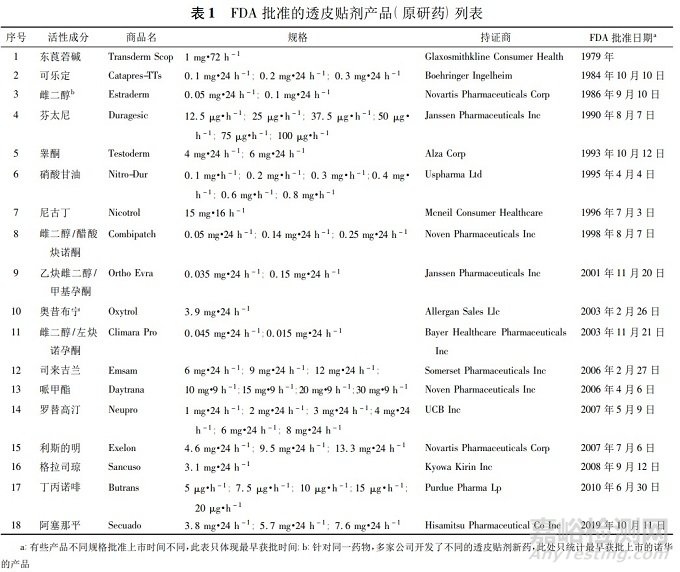
透皮贴剂一般由背衬层、含药骨架、压敏胶和保护层等数层组成,按照释药机制可以分为储库型和骨架型。储库型经皮给药系统是药物或经皮吸收促进剂被控释膜或其他控释材料包裹成储库,由控释膜或控释材料的性质控制药物的释放速率。骨架型经皮给药系统是药物溶解或均匀分散在聚合物骨架中,由骨架的组成成分控制药物的释放,示意图见图1。
欧美关于透皮贴剂的要求及实践
欧美关于透皮贴剂的一般要求
透皮贴剂规格的定义
为了保证透皮贴剂的活性成分在贴敷后的使用期间达到并保持预期的释药速率,保证入血的药量,进而保证疗效,透皮贴剂通常具有过量的载药量,即其实际载药量高于药物释放的总量,使用后的贴片中仍会残留一定量的药物。
据统计目前市售的TDDS在预期使用期后,会残留初始药物总量的10%~95%的药物。由于透皮贴剂需要药物过量并存在残留,采用透皮贴剂的载药量(所含药物总量)标示其规格是不合理的。
欧美均要求用释药速率(delivery rate,如mg/h,mg/24h)标示透皮贴剂的规格,如EMA《透皮贴剂质量研究指南》明确规定透皮贴剂的规格(dosage strength)定义为单位时间向体内释放活性成分的总量,通常为每24h向体内释放的药量。
以FDA批准的透皮贴剂为例(见表1),可见已批准的透皮贴剂均以释药速率(mg/h或mg/24h等)作为规格。
透皮贴剂规格的测定
与口服固体制剂的规格即载药量不同,透皮贴剂的规格通常是以其表观的释药速率表示(如每小时释放药物的量)。最常见的确定该速率的方法有:①通过人体药动学试验量化药物释放进入人体循环系统的量。②通过测定贴敷一定时间后贴片残留的活性成分的量,从而计算获得贴敷期间药物释放的量。
这些方法通过假设/估计获得表观药物释放速率。每种方法得出的规格结果都可能存在较大的差异性,2种测定方法所反映的规格量值也存在差异。
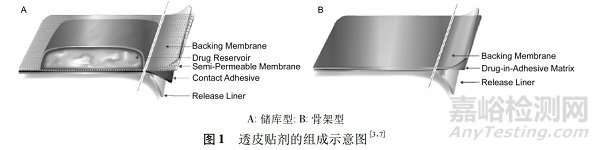
以利斯的明透皮贴剂原研药(商品名:Exelon)为例,在一项单中心、开放、平行、4周期、剂量递增的多次给药试验(试验代号:2331,随机招募51例53~84岁的患者,共30例完成试验)中测定了4种不同面积的贴片贴敷24h后的药物的残留量,进而计算释药速率(规格):释药速率=(载药量-残留量)/贴敷时间,并且结果显示平均约有50%的药物从贴片中释放出来(见表2)。
规格与载药量和贴片尺寸的关系
针对透皮贴剂规格标示方式的特殊性,为指导临床用药,说明书中需要明确每个规格的载药量和贴片面积。
通常,在FDA批准的新药的说明书中“12.3 Pharmacokinetics”项下交代药物从贴片中释放入人体的百分比,在“11 Description”或“16 How supplied/storage and handling”项下会列出透皮贴剂规格(释药速率)与载药量和贴片面积的对应关系。
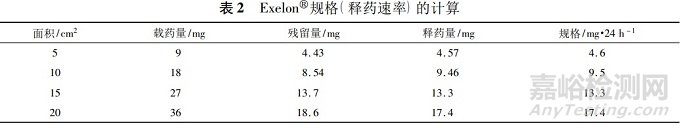
如FDA批准的罗替高汀透皮贴剂(商品名:Neupro)的说明书“11Description”项下描述的规格、载药量和贴片面积的对应关系见表3,“12.3 Pharmacokinetics”项下描述为“贴敷24h约45%的药物从贴片中释放出来”。
如FDA批准的利斯的明透皮贴剂(商品名:Exelon)的说明书“12.3 Pharmacokinetics”项下描述为“贴敷24h约50%的药物从贴片中释放出来”,“16 How supplied/storage and handling”项下描述的规格、载药量和贴片面积的对应关系见表4。
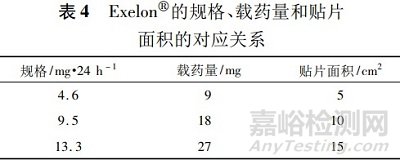
欧美关于透皮贴剂仿制药的技术要求
透皮贴剂仿制药规格、载药量和贴片面积活性的要求
仿制药须与参比制剂达到生物等效,对于透皮贴剂来说,要达到生物等效,必须保证仿制药从贴片中释放到人体的量与参比制剂一致,且贴敷时间与参比制剂一致,因此释药速率(释药量/贴敷时间)要与参比制剂一致。
EMA指南《缓释制剂的药代动力学和临床评价指导原则》中明确规定“透皮给药系统仿制药在单位时间内释放活性成分的量与参比透皮给药系统相同。此定义与一般定义不同,因为活性物质的总量可能不同,但仿制药与参比制剂在单位时间释放的活性物质的量应该相同”。
透皮贴剂仿制药开发的一个重要目标是所开发的仿制药可与参比制剂达到相同的释药速率,但其处方工艺并不一定要与参比制剂相同,因此透皮贴剂仿制药的载药量和/或贴片面积可能与参比制剂相同,也可能不同。
监管机构鼓励开发更小的贴片面积和更小的载药量(仿制药残留活性成分的量不应大于参比制剂)的仿制药,以尽可能降低残留物对环境的危害,除非有充分的理由和依据说明在获益/风险方面有所改善(例如改善皮肤耐受性、黏附性质、潜在结晶、冷流等)。
FDA批准的利斯的明透皮贴剂等的仿制药与相应的参比制剂的释药速率、载药量和贴片面积的对比(见表5),可以看出仿制药与参比制剂相比,释药速率均相同,而载药量和贴片面积可以不同。
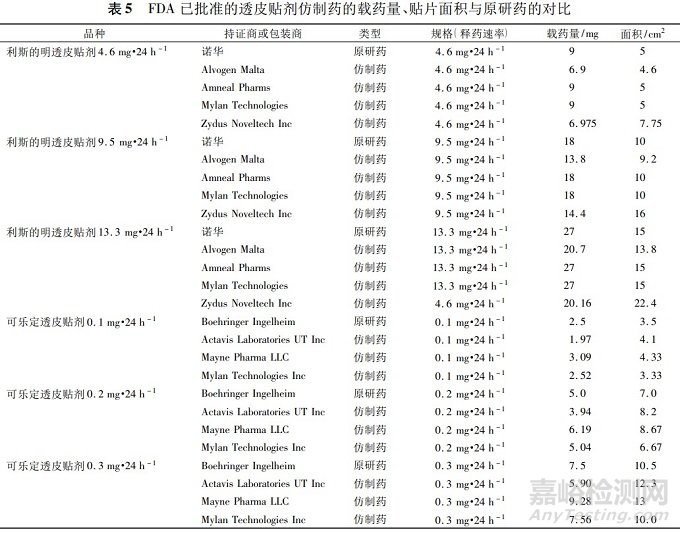
透皮贴剂仿制药体外释放度的要求
由于透皮贴剂的体外释放方法可能并未对体内性能进行模拟,因此,释放度方法只是作为质控方法,以确保上市销售的产品与关键临床批产品一致、上市销售的产品在货架期内释放度合格。
透皮贴剂常用的释放度方法为桨碟法、转筒法和往复支架法,每个品种具体的方法可以参考FDA溶出度数据库中相应的品种的方法或者美国药典(USP)中相应品种的方法。
为描绘释放曲线,每批通常要求最少提供12个单位(对于常规放行,可以接受不少于6个单位)的样品。对于透皮贴剂仿制药的释放度限度,应基于BE批、注册批(稳定性批)等批次的结果合理制定。
透皮贴剂仿制药临床试验的要求
FDA为指导仿制药开发,在其网站发布了一些特定产品的仿制药研究指南(Product-Specific Guidances for Generic Drug Development),经过检索,共有22个透皮贴剂品种(存在同一个药物开发出多个不同制剂的情况)已发布仿制药研究指南。
综合这类产品的仿制药技术要求,可以看出透皮贴剂仿制药要求开展3个人体试验,分别是:①单次、两序列,两周期、双交叉的生物等效性(BE)试验[药动学(PK)终点]。②单次、两序列、两周期、双交叉的黏附力试验。③随机、评估者盲法、受试者个体内重复的皮肤刺激和致敏试验。其中黏附力试验以及皮肤刺激和致敏试验可以单独开展,也可以与PK为终点的BE试验合并进行。
对于多规格产品,一般要求开展其中一个规格的BE试验,其他规格满足以下条件可以申请豁免:①一种规格的BE试验可接受。②所有规格体外溶出度试验可接受。③所有规格处方比例相似。
透皮贴剂仿制药的研发
透皮贴剂仿制药的制剂开发
与其他剂型的仿制药开发一样,透皮贴剂仿制药的开发要充分应用质量源于设计(quality by design,QbD)的理念。QbD的内容包括:确定药品关键质量属性的目标产品质量概况,生产设计和对关键物料属性的识别和理解,生产工艺设计和对关键工艺参数的识别和理解,控制策略包括对原料药、辅料和制剂的质量标准以及生产工艺的控制,工艺能力和持续改进。
开发透皮贴剂仿制药,首先要对参比制剂进行分析,包括临床使用、药动学特点、体外药物释放特点、理化性质、处方组成等方面,在此基础上进一步明确QTPP(见表6),在此基础上开发仿制药。

在制剂开发中要确定体内释药速率、活性成分的利用率(即活性成分从贴剂中释放出来的百分比)和活性成分的残留量,这些参数是影响制剂有效性和安全性的重要因素。另外,对于黏附性、皮肤耐受性、体外释放和皮肤渗透性以及贴片的尺寸、使用的方便性等也应当进行研究。
透皮贴剂的质量属性及质量控制
透皮贴剂的质量属性包括外观、鉴别、释放度、有关物质、残留溶剂、含量均匀度、含量、保护层剥离力、黏附力、微生物限度等,需要通过研究和风险评估来确定哪些质量属性为透皮贴剂的关键质量属性CQAs。
通常情况下,透皮贴剂的外观能够看出产品是否存在明显的质量缺陷,有关物质、残留溶剂和微生物限度影响产品的安全性,含量和含量均匀度影响产品的有效性和安全性,体外释放度反映透皮贴剂制造工艺的稳定性和均匀性,进而影响安全性和有效性,黏附力反映透皮贴剂与皮肤黏附的牢固程度,进而影响透皮贴剂的安全性和有效性,这些质量属性均属于CQAs。
透皮贴剂仿制药通常与参比制剂对比以下质量参数:规格(释药速率)、贴片中活性成分的载药量、单位面积的体内释药速率、活性成分利用率、贴片面积活性、残留量、使用说明、使用周期。除此之外,还要对比体外特性,包括药物释放、体外皮肤渗透和黏附性。对于仿制药来说,规格(释药速率)必须与参比制剂相同。
透皮贴剂注册批批量的要求
根据FDA指南《ANDAs:原料药和制剂稳定性试验问答》(ANDAs:Stability Testing of Drug Substances and Products Questions and Answers),对于透皮贴剂的批量,要求3批注册批中的2批至少为拟定商业批批量的1/10或25000个制剂单位(每种规格,取大值),第3批可以小于拟定商业批批量的1/10,但不得少于中试批批量的1/6。
对于骨架型透皮贴剂,以透皮贴片大小(表面积)来确定不同规格时,可采用3批骨架层生产的贴片研究数据,如适用,每批骨架层可以被裁切以支持多种不同规格的申报。
2018年6月22日国家药品监督管理局药品审评中心发布的《化学仿制药注册批生产规模的一般性要求(试行)》中对透皮贴剂要求:注册批3批均应至少达到拟定商业化生产规模的10%(包装后)或25000个制剂单位(每种规格),两者中选更多的。对于骨架型产品,以透皮贴片大小(表面积)来确定不同规格时,建议申报时提交采用3批骨架层生产的贴片研究数据。
我国透皮贴剂监管目前存在的问题及思考
关于透皮贴剂的规格
我国并未明确规定透皮贴剂的规格的定义,《中华人民共和国药典》2015年版二部中仅收载了雌二醇缓释贴片,其规格标示为:2.5mg(4.0cm×2.6cm),该规格标示是不准确、不科学的。笔者调研发现国内批准上市的透皮贴剂的规格标示形式未统一,进口品种中外规格标示不一致,举例如下(见表7)。

由表7可见,欧美国家上市的透皮贴剂规格均以释药速率标示,但国内已上市透皮贴剂的规格标示方式混乱,有以总载药量、贴片面积、释放速率作为规格,或取其中两者作为规格等多种标示方式。
我国透皮贴剂规格无定义、标示方式不统一,会给监管带来很多问题:①不同的进口原研品种国内规格标示混乱。②按照仿制药要与参比制剂规格一致的要求,如果要仿制这些进口原研制剂,国内仿制药企业必须做到规格一致,这将增加不必要的技术障碍,不利于我国仿制药的研发和上市。③对于我国已有仿制药的品种,在开展一致性评价时,将遇到无法找到合适参比制剂的问题。
为解决这些问题,首先要明确和统一我国透皮贴剂规格的定义,建议参考欧美实践,以“释药速率”标示规格,可以在《中华人民共和国药典》贴剂通则中予以明确。
关于透皮贴剂仿制药的技术要求
因透皮贴剂的复杂性,FDA将其归为复杂剂型的复杂产品。针对透皮贴剂及其仿制药,欧美均制定了相应的技术指导,指导企业研发和注册审评。我国目前除了《中华人民共和国药典》2015年版四部贴剂通则外,无针对透皮贴剂研发的技术指南,建议我国参考这些指南,制定我国透皮贴剂的技术指导原则,并明确透皮贴剂仿制药要求规格(释药速率)与参比制剂一致,而载药量和贴片面积可以与参比制剂不同。另一方面,建议针对具体品种,制定相应的仿制药研究指南。
关于透皮贴剂仿制药的说明书
为规范和指导临床用药,首先明确要求透皮贴剂产品在说明书的“用法用量”项下注明剂量,用表格的形式表述每种规格相对应的贴片面积和载药量。其次,对于透皮贴剂仿制药,可以参考参比制剂的说明书撰写透皮贴剂仿制药的说明书。
如前所述,透皮贴剂仿制药的规格(释药速率)与参比制剂相同,而载药量和贴片面积与参比制剂不同,在仿制药说明书中要体现载药量和贴片面积。此外,加强对医生、护士和患者的教育,使其加强对说明书中规格(释药速率)、载药量和贴片面积之间关系的认知,以便在说明书的指导下合理用药。
结语
透皮贴剂不同于口服固体制剂,在监管过程中要根据其特点制定相应的监管要求。国外尤其是美国透皮贴剂临床应用已有40多年的历史,虽然规格的定义和标示与其他剂型不同,但在说明书的指导下使用并未造成临床使用和监管混乱。
对于特定的产品,在研究过程中已经确定了活性成分的利用率,即从贴片中释放出来的活性成分的百分比,在这个前提下,在质量标准中控制含量(载药量),就能保证活性成分由贴片中释放至人体的量,进而保证了疗效和安全性。
我国规范透皮贴剂的规格、说明书及仿制药技术要求,将有利于促进我国透皮贴剂的研发、提升仿制药质量疗效、提高药品供应保障能力,也有利于透皮贴剂的监管,更好地满足临床用药及公共卫生安全需求。

来源:《中国新药杂志》


