在生物类药物生产管理中,制剂生产管理至关重要,其中无菌生产管理更是核心环节。本文从生产工艺特性、环境控制、生产管理及无菌控制、质量监控等方面,阐述了在GMP体系下确保生物制药制剂无菌生产的具体措施,以保障药物生产的安全性、有效性和质量一致性。
基于GMP体系,生物制药的制剂生产管理显得尤为重要[1-2]。通过实施GMP标准,企业能够确保产品的安全性、有效性和质量一致性,进而提高患者的用药安全。
(一) 生物制药制剂生产工艺特性
生物制药制剂的生产工艺特性涉及多个方面,包括生产工艺的复杂性、质量控制要求及特定的环境和设备要求等。
1.1 工艺复杂性
技术手段多样:通常采用基因工程、细胞培养、发酵等生物技术手段。如基因工程药物是通过重组DNA技术将目标蛋白表达基因植入宿主菌(细胞)中,再进行大规模发酵生产。
流程环节众多:一般包括原料的选择、原料的预处理与保存、生物催化剂的制备、设备与培养基的灭菌、无菌空气的制备、发酵及工艺控制、产物的分离纯化、成品的检验与包装等环节。
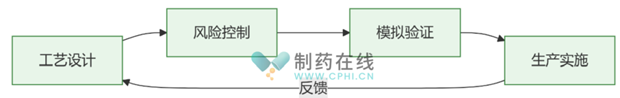
生物制药制剂生产工艺流程(图片来源:作者绘制)
1.2 质量控制严格
检测指标多样:除了常规的理化检验指标外,更要有生物活性检验指标,如电泳法检测蛋白质纯度、HPLC法检测物质纯度和肽图、细胞法、病毒法检测物质的活性特征等。
各环节严格把控:每一个生产步骤都需要进行严格的检测,以确保产品的纯度、安全性和有效性。例如在制剂生产中,要对原液的配制、分装、贮存等过程进行微生物限度检测等。
1.3 无菌生产要求高
生产环境洁净:大多数生物制药产品需要在无菌环境下生产,通常在洁净室内进行,不同级别的洁净区有不同的要求,如A级洁净区属于高风险区域,如灌装和无菌配制区,其清洁标准极高。
采用无菌技术:采用无菌过滤、灭菌技术等,如除菌过滤作为一种常见的微生物控制手段,能够有效去除液体中包括细菌和支原体在内的微生物。
1.4 温度和环境控制精准
温度敏感:生物制药产品中的活性成分对温度极为敏感,如许多蛋白类药物在高温条件下容易发生变性而失活,因此在生产过程中需要精确控制温度,以保持产品的活性。
其他环境因素:pH值、氧浓度等环境因素也需要精确控制,以确保产品的稳定性和质量。
1.5 稳定性和储存要求特殊
配方工艺复杂:生物制药制品通常需要复杂的配方工艺以提高其稳定性,如添加合适的辅料、调节pH值等。
特定储存条件:一般需要在特定的条件下储存,如冷藏或冷冻,以防止产品变质或失效。
1.6 生产成本较高
设备投入大:需要大量的专业设备和先进的技术,如发酵罐、纯化设备、冻干机等,这些设备的购置和维护成本较高。
质量控制成本高:由于生产工艺复杂且质量控制要求严格,需要进行大量的检测和验证工作,增加了生产成本。
(二) 生物制药制剂生产环境控制
2.1 洁净区划分
根据生产工艺和产品要求,将生产车间划分为不同级别的洁净区,如A级、B级、C级、D级等。不同洁净区之间应有合理的气流组织和压差控制,防止空气交叉污染。
2.2 空气净化
采用高效的空气净化系统,如HEPA过滤器、紫外线灭菌器等,对洁净区的空气进行过滤和消毒。定期对空气净化系统进行维护、检查和验证,确保空气洁净度符合要求。
2.3 温湿度控制
生物制药制剂生产对温湿度要求较为严格,一般温度控制在18-26℃,相对湿度控制在45%-65%。通过空调系统对洁净区的温湿度进行控制和调节,定期对温湿度监测设备进行校准和维护。
(三) 无菌制剂生产管理及无菌控制
所有从事无菌药品生产的人员都应经过培训,包括生产操作人员、生产辅助人员等。除此之外,还需要重点关注物料与耗材管理、设备管理、厂房设施管理、无菌工艺管理等方面内容。
3.1 物料与耗材管理
质量标准与放行检测:无菌药品生产的相关原辅料必须有相应的微生物限度控制标准和细菌内毒素质量标准。每批次物料在放行检测过程中,微生物指标需符合要求后才可用于生产。对于无菌制剂生产的包装材料,尤其是内包装材料,如果是即用型包材,不但要严格审核其质量标准,而且要确认其达到质量标准所采取的控制手段及措施。
使用前处理与控制:对于物料,使用前需要确认放行检测结果是否符合标准、包装是否完好、取样是否具有代表性。对于包材,尤其是内包材,如果采用的是即用型包材,使用前需要核对其成分分析报告,确认其放行检测结果符合质量标准,此外,使用前还需确认其包装的完整性。对于与药品直接接触的关键耗材,如果要进行清洗、灭菌,需要进行清洗效果确认或验证。
3.2 设备管理
选型与设计:在设备选型时,要从产量、生产产品种类、后期运维及能耗等角度出发确定大方向,同时充分考虑后期生产的无菌保障。在设备进行设计确认过程中,需要充分考虑人员操作的便捷性,还要考虑对A级区等关键区域的气流影响。
维护与维修:对于无菌制剂生产设备,出现异常时,需由经过专业培训的设备工程师进行维修。维修时要遵循相应的GMP规范,尽量通过手套或已灭菌的工具进行相应维修,维修过程动作要轻缓,减少对气流的阻挡。维修结束后,需要对维修区域及其周围区域进行相应的清洁消毒,清洁消毒完成后,还要等A级区环境进行自净,自净结束后才可恢复生产。
3.3 厂房设施管理
布局:厂房设施布局要根据药品生产中发挥的功能进行分区,不同功能区域要求布局合理,既要减少人员、物料的交叉,又要防止不同功能区域的污染及交叉污染。对于不同洁净级别的功能区,其压差至少要大于等于10Pa;对于同一洁净级别的不同功能区,也应设置一定的压差梯度,通常设置为5Pa。
设计:厂房设计时要考虑日常监控和参观的需求,尽可能多设置观察窗、参观走道等。从低级别区域进入高级别洁净区宜设置缓冲间,尽量避免跨级别进入。跨洁净级别的管路需要做好密封,跨洁净级别的传递窗、缓冲间需要监控压差。在满足工艺需求的前提下,尽量减少洁净区用水点,洁净区要减少地漏的布置,下水口要密封,需要设置空气阻断装置防倒灌。
3.4 无菌工艺管理
除菌过滤及无菌灌装工艺设计:对于生物制剂,一次性技术的应用越来越广泛,尤其是对于除菌过滤及无菌灌装工艺来说。一个好的设计通常要考虑系统无菌性能、简单便于操作、药液损耗低等因素。在满足上述条件的基础上,选择合适的一次性耗材厂家,根据自身设备特点,和供应商一起设计符合自身无菌工艺要求的一次性系统。
无菌工艺模拟:无菌工艺模拟是验证无菌生产工艺是否可靠的最直接最有效的方式。在无菌工艺模拟实施前,先要完成风险评估,包括无菌工艺模拟干预情况评估、无菌工艺模拟时限考察评估、无菌工艺模拟环境菌选择、无菌工艺风险点识别与评估等。实施过程中需要模拟最差条件,如最大最小灌装速度,物料灭菌后存放的最长时间等。
无菌制剂生产管理流程(图片来源:作者绘制)
(四) 生物制药制剂生产质量监控
4.1 微生物监测
定期对洁净区的空气、表面、人员手部等进行微生物监测,包括沉降菌、浮游菌、表面微生物等检测项目。根据监测结果,评估洁净区的微生物污染水平,及时发现和解决微生物超标问题。
对无菌制剂产品进行无菌检查,采用薄膜过滤法、直接接种法等方法,检测产品中是否存在微生物。无菌检查应在符合要求的无菌检查室进行,检查人员应经过专业培训,熟悉无菌检查的操作规程和质量标准。
4.3 热原与细菌内毒素检测
生物制药制剂中的热原和细菌内毒素可能引发患者的不良反应,因此需要对产品进行热原与细菌内毒素检测。采用鲎试剂法等方法进行检测,确保产品中的热原和细菌内毒素含量符合质量标准。
生物制药制剂生产质量监测级方法(图片来源:作者绘制)
(五) 总结
在GMP体系下,确保生物制药制剂的无菌生产是一个系统工程,需要从人员、设备、环境、工艺、质量监控等多方面进行严格管理和控制。只有将这些环节有机结合起来,才能有效降低微生物污染风险,确保生物制药制剂的质量和安全性,为患者提供安全有效的药品。
参考资料:
[1] Sampathkumar K, Kerwin BA. Roadmap for Drug Product Development and Manufacturing of Biologics. J Pharm Sci. 2024 Feb;113(2):314-331.
[2] EU-GMP, Annex 2B Manufacture of biological medicinal substances and products for human use.
作者简介:Sophia,主要从事生物医药行业发展研究、药物科普等方面工作。