原料药中间体的质量研究和控制策略是确保最终原料药质量的关键环节,其目的是通过科学的分析和控制手段,确保中间体符合既定的质量要求,从而保证最终原料药的安全性、有效性。
本文主要从以下3方面阐述如何开展中间体的质量研究:
● 国内外法规(申报资料) 要求汇总
● 质量研究的一般要求
● 质量研究后的一般输出
1. 定义:ICH Q7 和Q3A(R2)的术语部分,均给予了中间体的定义如下:
注:湿品(待干燥得API)以及干燥后待粉碎的API,根据上述的定义,严格意义是不属于中间体范畴,干燥的IPC(水分或者LOD)来代替中间体的控制。
01申报资料的法规要求汇总
中间体需要递交的研究内容,主要来源于ICH M4Q 3.2.S.2.4中如下要求:
3.2.S.2.4 关键步骤和关键中间体的控制(名称、厂商)
Intermediates: Information on the quality and control of intermediates isolated during the process should be provided.
中间体:应提供在生产工艺中分离的中间体的质控信息。
上述M4Q主要为框架式要求,更为详细的还需看如下一些法规和指南3.2.S.2.4的要求:
a) 总局关于发布化学药品新注册分类申报资料要求(试行)的通告-2016年80号文
3.2.S.2.4关键步骤和中间体的控制
列出已分离的中间体的质量控制标准,包括项目、方法和限度,说明标准制定的依据,对关键中间体的主要质控方法(如杂质控制方法),应提供必要的方法学验证资料。
明确反应副产物和副反应产物的产生及控制方法、限度、数批样品的检测结果与图谱。若涉及引入新手性中心的反应,应详细提供异构体杂质的分析方法与控制策略。
b) 药审中心关于发布《境外已上市境内未上市化学药品药学研究与评价技术要求(试行)》的通告(2021年第21号)
对于已分离的中间体,申请人应制定包括检测项目、分析方法和可接受标准的质量标准,并说明质量标准制定的依据。关键中间体的主要质控方法(如杂质控制方法)应进行包括专属性和灵敏度等的方法学验证。申请人应根据杂质转化和清除研究结果,为原料药过程控制提供合理依据.
c) 关于公开征求《已上市化药药品补充申请药学自评估报告(原料药/制剂)(征求意见稿)》意见的通知2024年7月26日
中间体的控制:结合原料药整体质量控制策略,简述中间体的质量控制策略,包括杂质分析、内控标准(包括项目、检测方法和限度)、样品检验等,并简述必要的方法学验证。
如中间体存在暂存情况,简述中间体暂存条件和时间的验证情况。
d) [1]EDQM: Content of the dossier for CEP applications for chemical purity and microbiological quality of substances for pharmaceutical use (2024)
Controls of critical steps and intermediates (3.2.S.2.4):
A suitable and detailed specification is expected for each isolated intermediate, along with analytical methods descriptions. The specification should generally be justified and information on the impurities found in isolated intermediates during manufacture should be included (e.g. specified, unspecified or total impurities) as necessary.
每个分离出的中间体均应提交适当的详细的质量标准,以及分析方法描述。质量标准一般应经过论证,必要时还要包括生产过程中分离出来的中间体内的杂质的信息(例如,特定杂质、非特定杂质或总杂)。
e) [2]EMA: Guideline on the chemistry of active substances(EMA/321776/2024)
Control of Critical Steps and Intermediates 3.2.S.2.4
Intermediates: 中间体
Information on the quality and control of intermediates isolated during the process should be provided. If non-compendial methods are used to control the intermediate, they should be suitably validated.
应提交在工艺中被分离出的中间体的质量和控制的信息。如果使用了非药典方法来控制中间体,则这些方法应进行适当验证。
Validation data is not expected unless the test in question is essential for the control strategy of the active substance (e.g. removal of a mutagenic impurity). Information on the characterization of these intermediates should be provided (Ref 7).
如果这些检验方法并不是活性物质控制策略(例如,除去诱变性杂质)的重要部分,则其验证数据不需要提交。应提交这些中间体的特性资料。
If an intermediate in the proposed synthesis of the active substance is itself an active substance covered by a monograph of the European Pharmacopoeia (Ph. Eur.) covered by a valid CEP, then the CEP can be submitted as an alternative to submitting its process description. Documentation on the additional chemical transformation steps from the intermediate to the active substance should be provided in 3.2.S.2.2. The manufacturers involved in the process covered by the CEP should be listed in module 3.2.S.2.1 and the QP declaration (Ref 13).
如果所拟活性物质的合成中所用的某个中间体本身是一个活性物质,已载入EP各论并且具备有效CEP,则可以提交CEP替代提交工艺描述。在3.2.S.2.2中应提交从该中间体开始到活性物质的其它化学转化步骤的文件资料。应在模块3.2.S.2.1和QP声明中列出CEP中所述工艺所涉及的生产商。
If an intermediate in the proposed synthesis of the active substance is itself an active substance already included in a finished product authorised in the EU and documented in an ASMF or in module 3, then this can be referenced. Complete information on the manufacturing process (3.2.S.2), starting with the starting materials will still need to be submitted, either as part of a new ASMF or in the dossier.
如果活性物质的合成中所用的某个中间体本身是一个活性物质,该活性物质已经用于EU批准的某个制剂中,并且在一份ASMF或模块3中有载录,则可以进行引用,但仍需提交从中间体开始的完整的生产工艺信息(3.2.S.2),可以是作为新的ASMF的一部分,也可以是写在注册文件里。
f) [3]FDA: GDUFA 要求下 II 类原料药 DMF 完整性评估
3.2.S.2.4 Contains information for Controls of Critical Steps and Intermediates, as follows:
29. Specifications for each identified intermediate. 每个识别的中间体的质量标准。
30. Analytical methods for each intermediate.每个中间体的分析方法。
因为美国的DMF Type II中是包含:Drug Substance, Drug Substance Intermediate, and Material Used in Their Preparation; or Drug Product.
有很多中间体的生产商也会递交中间体DMF(Secondary DMF)供原料药厂家DMF(Primary DMF) 的引用。那么中间体的质量研究不充分,势必会影响DMF的审评。
综上所述,总结在申报资料里需要递交哪些内容涉及到质量研究和控制策略:
1. 中间体的质量标准
2. 中间体的质量标准的制定依据
3. 中间体的分析方法以及方法验证
4. *中间体的结构确证(EMA对ASMF的申报要求)
5. *中间体的稳定性研究(如有暂存情况)
*:特殊要求
02质量研究的一般要求
中间体涉及的质量研究内容比原料药和SM要少一些。因为SM是外购为主,其不可控制性远不如中间体。
下文将结合国内外监管机构发中常见的一些发补案例,展开阐述:
1. 中间体的质量标准
通常设置的项目有性状,鉴别,水分/干燥失重,无机盐类(炽灼残渣),有关物质,异构体,致突变杂质,含量。
2. 中间体的质量标准的制定依据
中间体的质量标准,不像SM有外购工厂提供的一些COA等信息可以参考。
一般根据其对后续反应及原料药质量的影响,来制定相关的质量标准。
主要从以下几个方面,阐述:
● 性状以及理化项目
● 鉴别
● 无机杂质
● 水分或干燥失重
● 有关物质
● 致突变杂质
● 其他杂质(残留溶剂,元素杂质,亚硝胺杂质)
2.1 性状以及理化项目
性状(如外观、颜色、物理状态)、理化项目(熔沸点、比旋度、pH等)。
一般根据实际的中间体的情况制定。通常因中间体需要后续继续投料,考虑时间问题,理化项目可以减少不订。
2.2 鉴别
案例1(FDA):We acknowledge you have provided intermediate specifications for XXX and XXX In S.2.4, However, neither of them has identification tests. Please include
orthogonal identification tests in your intermediate specifications.
已在S.2.4中为XXX和XXX提供了中间体质量标准,但两者都没有进行鉴定测试。请在中间质量标准中包括正交鉴定试验。
案例2(EMA):Identification of crude API solely by a single chromatographic retention time is notregarded as being specific and should be supplemented with a further method, e.g. IR
仅通过单一色谱保留时间鉴定API粗品不被视为具有特异性,应补充进一步方法,如IR
总结:通常要求有2个交叉引用的鉴别项目:IR,UV,HPLC,理化(颜色反应,沉淀)是常用。
2.3 无机杂质
生产工艺中,经常会使用到大量的无机盐类,同时各化学反应也会生成很多无机类的副反应。那么得到中间体通常并不是单一物质,有可能是含有大量的无机盐的混合物。
是否需要对无机杂质类似于API中那样进行严格控制?
需要结合API工艺的清除情况,无机杂质的限度可以制定更高。
后续反应可能会有很多洗涤操作,会去除部分无机盐类,其限度比原料药中要求会高的多。
考察到对于后续投料,要求检测不需要太长时间,所以往往不需要非常精确的定量(ICP-MS,AAS方法)。
快速的粗略定量,往往是符合工业生产要求,其实也有助于后期的投料定量,避免其他物料的过量投料,造成不必要的浪费,也有利于收率波动时候的调查。
2.4 水分或干燥失重
因原料药生产经常会连续投料,那么中间体也会以湿品形式,往下投料。
或者一定程度的干燥,这和原料药成品对水分(或干燥失重)的严格要求(不得过0.1%等等)是不一样的。
因湿品往往无法定量投料(含水或者含溶剂),设置水分或者干燥失重,有助于进行定量投料,减少不必要的过量投料。
如果后续反应对水分有要求,诸如:格氏反应、有机锂试剂反应、金属还原反应、傅克反应、Suzuki偶联),那么还是需要对水分进行严格控制的。
2.5 有机杂质
关于有机杂质的质量研究,我们从下面2个案例,给予说明:
案例3(CDE):
① 请结合制备工艺,进一步完善杂质分析,补充进行如杂质XXX、二聚体等的研究(如采用MS方法)。请根据多批检测结果和追踪研究结果,拟定合理的限度;
② 建议将检出较大的杂质按特定杂质单独控制,并请收紧其它单杂的限度,以保证中间体质量的可重现性;同时请补充提供方法学验证资料。
③ 杂质YYY为主要的工艺杂质,请提供其结构确证资料,该杂质带有苯胺警示结构,请参照ICH M7采用两个原理互补的权威软件对杂质YYY的致突变进行预测,若为潜在致突变杂质,请参照ICH M7 进行研究和控制;另该杂质带MMM基团,可进一步参与反应,请提供转换杂质的研究情况;
④ 杂质MMM和杂质NNN的校正因子较大,请加校正因子定量;
通过上述补充资料要求可以看得出:
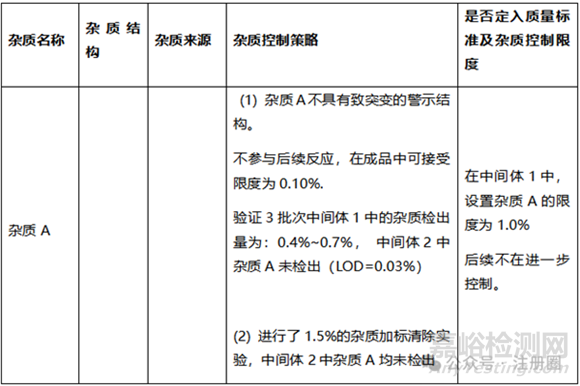
(1) 中间体中需要补充额外的杂质研究:因杂质分析不全面,诸如聚合物杂质。
(2) 需要关注:带有反应基团,可参与后续反应的杂质残留情况。
(3) 杂质的定性:含警示结构,需要按M7致突变杂质研究。
(4) 含量较大的杂质需要定性,单独控制
那么中间体中有机杂质的质量研究的思路:
● 综合分析杂质的来源和去向
来源分析:反应的副产物,原料的残留,上一步杂质的携带。
结合反应机理和文献,对中间体中实际检出的杂质进行定性(可采用MS),建立中间体的杂质谱。
根据多批次的杂质的残留水平和去向,进行杂质的定量研究,开发相关的分析方法,制定相关的内控标准。
那么研究的重点在于杂质结构的确定和去向。
● 分析方法
中间体的分析方法也是需要通过验证的,验证后可得到各杂质的校正因子,来进一步确认计算是否需要加校正因子。
校正因子: CDE电子期刊中《HPLC法校正因子研究中的几个问题》中提到:一般情况下,校正因子在0.9~1.1时可不予校正,直接采用不加校正因子的自身对照法定量;超出该范围,如采用主成分自身对照法的定量方式,须用校正因子进行校正,即“加校正因子的主成分自身对照法”以保证杂质定量的准确性;
2.6 致突变杂质
中间体中的致突变杂质研究和控制,是很常见的,尤其是使用ICH M7中的option3.即:
在中间体的质量标准中对杂质进行检测或进行过程控制,制订一个高于原料药中该杂质可接受限度的标准。
对低限度的检测,往往依赖高灵敏度的检测机器(诸如:LC-MS/MS, GC-MS/MS),这种对于生产型企业是不适用的。按照一般杂质限度的控制,更符合企业的实际情况。
实际操作过程:
(1) 确定杂质限度: 按照对应的TD50或TTC 计算。
(2) 中间体中多梯度的限度(一般杂质限度)进行加标做清除试验.
(3) API结果检测:低于30%TTC,需要开发高灵敏度的分析方法(可以委外进行方法的开发和检测)
2.7 其他杂质(元素杂质,残留溶剂,亚硝胺杂质)
元素杂质和残留溶剂,尽管生产中可能会使用到各种溶剂和金属添加剂(催化剂等),但中间体作为一个过渡态,往往不在中间体中研究,重点体现在成品原料药中研究。
3. 中间体的分析方法以及验证
中间体和起始物料一样,均需要提供方法学验证资料。
上述案例中,验证可以表明杂质的校正因子。
中间体中当成特定杂质,需要进行定量验证;其他一般杂质或者可能存在的杂质,一般定性验证即可,可以减少部分的工作量。
4. *中间体的结构确证(EMA对ASMF的申报要求)
国内对中间体的结构确正,往往不做要求。但是对于欧洲ASMF来说,欧洲的各大监管机构,尺度并不统一。
案例5(EMA):Characterization data of INT-1 &INT-2 and their impurities through different techniques are missing and should be provided.
分析:API的结构确证主要有:
元素分析, 紫外, 红外, 核磁共振(1H-NMR、13C-NMR,COSY,HSQC,HMBC);
质谱;立体结构(单晶X-射线衍射);热分析(TGA &DSC);晶型(XRPD)。
那么中间体是否也需要做这么齐全呢?
答案是不需要:
简化版的结构确证,已经较为充分。
- 通过IR、NMR、MS、元素分析等手段,确认中间体的化学结构。
- 对于手性中间体,需明确立体构型(如手性HPLC, NMR等)。
5. *中间体的稳定性研究
案例6:请明确各中间体存放条件和时限,并提供确定依据。
对于非连续批次以及亚批混合成大批的情况,往往会存在中间暂存的情况。
也有些生产商将中间体进行售卖,都要进行稳定性研究,来确定暂存时限。
03质量研究后的一般资料输出
在申报资料中往往需要输出以下内容:
(1)中间体1
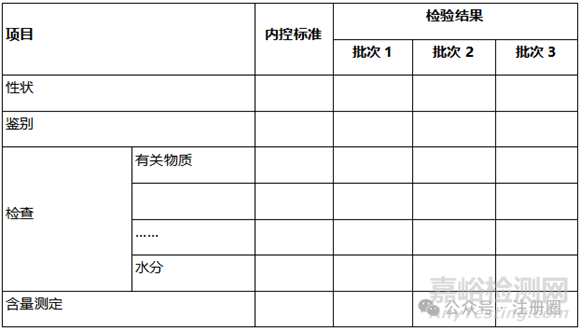
① 中间体内控标准与检验结果。
② 杂质分析:
③ 分析方法及方法验证:
主要控制项目(如有关物质、异构体、残留溶剂、含量测定等)的方法验证总结。
参考文献:
[1] Content of the dossier for CEP applications for chemical purity and microbiological quality of substances for pharmaceutical use:2024),
[2] Guideline on the chemistry of active substances: EMA/321776/2024)
[3] Completeness Assessments for Type II API DMFs Under GDUFA