您当前的位置:检测资讯 > 监管召回
嘉峪检测网 2019-05-15 16:09
前言
儿童药市场巨大,潜在规模1000亿以上,卫计委也多次发文鼓励研发儿童药。2018年初,CFDA上了新闻联播,强调儿科药、创新药。想必在不久的将来,儿童药的红利就要来了,尽管目前国内的政策、法规还不成形,但我们可以参考美国的。所谓他山之石,可以攻玉,转发科睿唯安权威文章一篇,希望对广大朋友有所帮助。
by Albane d'Argent /中文翻译:付倩
简介
五年前的2012年7月,美国国会通过了《美国食品药品监督管理局安全及创新法案》(FDASIA),其中将《儿科研究平等法案》(Pediatric Research Equity Act, PREA)和《最佳儿童药物法案》(Best Pharm-aceuticals for Children Act, BPCA)对儿科药物研发的影响定为永久性法律。在此法律基础上,美国食品药品监督管理局(FDA)采用胡萝卜加大棒——软硬兼施的策略,促进儿科药物研发和鼓励将儿科人群纳入临床试验。
在PREA和BPCA立法前,大多情况是将成人药品超说明书用药,给儿童使用。与大众的看法相反,儿童并不是缩小版的成人,通过体重的推算减小剂量的方式并不能确保药物的安全性和有效性。对临床试验中缺乏儿科人群的可能原因做出了一些假设。通常认为对此类弱势人群做进一步研究的重大障碍包括伦理学考虑、入组前获得儿童及其父母的知情同意。缺少儿科人群临床试验的其他原因包括:
儿童生长发育迅速,在试验开始到结束的过程中,这一特点增加了 试验结果评价的复杂性。
儿科人群被分段,导致需要对其进行不同的试验:新生儿与10岁儿童不具有等同性。FDA的指导原则——“PREA实施细则”中将0-16岁的人群分为四大类:新生儿、婴幼儿、儿童和青少年。
血样采集有严格的限定条件,这增加了儿科人群的血样采集的挑战性。
虽然方式不同,但PREA和BPCA都起到了促进效果,使包含儿科人群的临床试验数量增加。PREA相当于“大棒”,要求申办方在每份给FDA的申请中都提交儿科研究计划。除非经FDA同意准予豁免,所有新药临床试验申请均需提交儿科研究计划,即使此产品声称的适用人群仅为成人。儿科研究可推迟至批准后进行,这样药品在满足应用于成人的条件下,无需等儿科人群临床试验结果即可上市。申办方应在截止日期前向FDA提交儿科评估报告。联邦食品、药品和化妆品法案(the Federal Food, Drug,and Cosmetic Act, FD&CAct)中的505B(d)(1)部分,要求FDA给未完成下述内容之一的申办方发PREA不符合信:
1)按PREA的要求在截止日期前提交儿科评估报告;
2)申请或获准延期;
3)按要求递交儿科处方申请审批;
BPCA则作为“胡萝卜”激励申办方进行儿科人群临床试验。按BPCA的规定,对可能有利于儿童健康的药物,FDA可书面通知申办方进行儿科人群临床试验。此通知为非强制性要求,申办方可自愿选择是否开展试验。若申办方按FDA提出的要求进行研究,将依照FD&CAct中的505A部分获得6个月的市场独占期。也可应申办方的要求使用BPCA:对于需按要求进行PREA的药物,申办方可向FDA提交进行儿科研究的要求,用以接收书面的通知并合法取得6个月的市场独占期。自2007年9月27日签署美国食品药品管理法修正案(Food and Drug Administration Amendments Act, FDAAA)以来,FDA共做出了85个儿科药物市场独占期的决定。
对在美国进行了儿科人群临床试验进行多方面分析。以2015年为例,首先识别出2015年FDA批准的化学药品和生物制品新活性(novel actives ubstances,NASs)。其次对这些NASs的下述内容进行分析:被要求进行的儿科研究计划;被同意全部豁免或部分豁免的申请人,及其相应的被豁免理由。此外还分析了哪些治疗领域被要求进行儿科研究计划,以及哪些被全部豁免。得到的结果能辅助判断美国儿科研究的潜在趋势。表1为筛选出的供分析的申请。
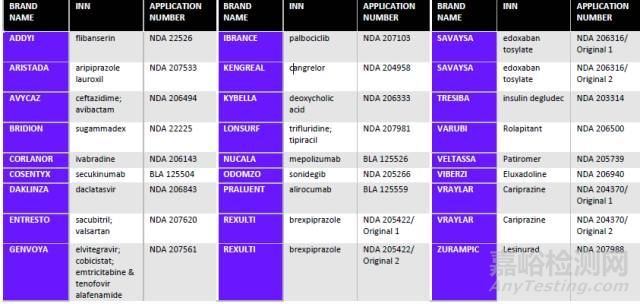
表1 用于本研究的产品:2015年FDA批准的新活性物质
来源:科睿唯安Cortellis 药政法规数据库:美国法规情报报告--儿科研究和发展综述
豁免
2015年FDA批准的27个NASs中,获得全部豁免、部分豁免和未获豁免的数量分别为13、9和5,占比分别为48%、33%和19%(见图1)。
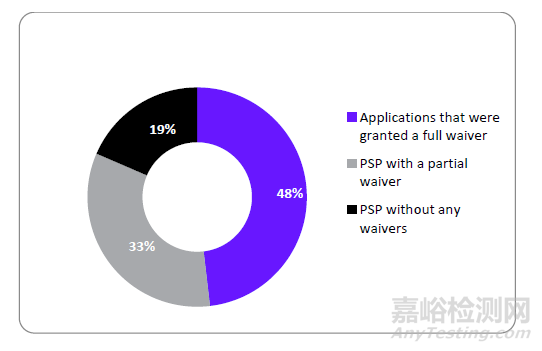
图1 2015年FDA批准的NASs儿科研究计划
(Pediatric Study Plans , PSP)豁免情况
来源:科睿唯安Cortellis 药政法规数据库:美国法规情报报告--儿科研究和发展综述
当FDA认为儿科人群临床试验是不切实际或不必要时,可全部或部分豁免儿科评估。获得豁免后,申办方无需符合儿科研究的要求。FDA可在一开始或后续按申办方的申请准予其豁免。
申办方满足下述条件之一时,所有儿科人群研究可全部豁免:
1.儿科评估的进行是不可能或不切实际的,如儿科患者数量过少或散布在不同区域;
2. 有明确的证据显示药品或生物制品对所有儿科人群无效或不安全;
3. 相较现有疗法,药品无法提供更有意义的疗效,不致适用于多数儿科人群。
2015年获得全部豁免的13个申请人中:11个的豁免原因是“儿科评估的进行是不可能或不切实际的,如儿科患者数量过少或散布在不同区域”(条件1);1个的豁免原因是“相较现有疗法,药品无法提供更有意义的疗效,不致适用于多数儿科人群”(条件3);还有1个获得全部豁免的同时满足条件1和3。没有因符合“有明确的证据显示药品或生物制品对所有儿科人群无效或不安全”(条件2)而豁免的申请。
对特定年龄段的儿科人群可给与部分豁免,豁免条件为前面全部豁免的条件加上下述第4条:
4. 无法给特定儿科人群提供其所需要的制剂。
大部分情况下,申请人获批的部分豁免仅针对年龄小的儿科人群,而对年龄大的儿科人群中仍需进行研究。2015年,所有的部分豁免都包含新生儿,但不包含12岁以上儿科人群,9个获批的部分豁免针对的人群为0-12岁(见图2)。
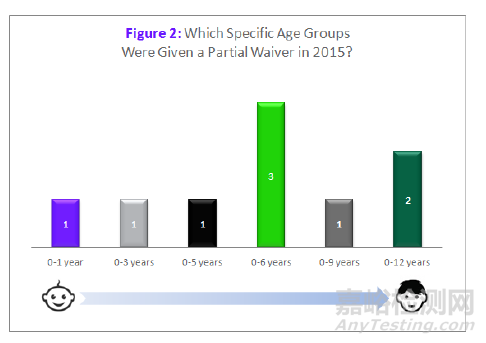
来源:科睿唯安Cortellis 药政法规数据库:美国法规情报报告--儿科研究和发展综述
无论全部还是部分豁免,都被申请人视为免于进行更多儿科临床试验的“圣杯”。尽管豁免的申请人众多,获批却并不容易,申请人必须向FDA提供支持性的文献证明申请的药品不适用于儿科人群。BPCA和PREA这两个法律通过胡萝卜加大棒的政策,达到了逆转以往趋势的目的。不仅使儿科人群评估不再能避免,同时通过给与6个月的市场独占期,使儿科人群临床试验的投资有合理的回报。
在部分申请人希望完全避免大规模儿科人群临床试验的同时,另外一部分申请人在尝试通过获批延缓来推迟试验。
豁免可免于进行儿科评估,延缓可推迟提交儿科评估。获准延缓后,申请人仍然需要提交儿科评估,只是需要选择最合适的时机来提交。FD&CAct的505B(a)(3)部分指出:“由FDA提出或申请人申请,FDA有可能批准延缓提交全部或部分评估……直至药品批准后的某个日期”。
可延缓的原因为:
5.进行儿科评估将延误成人患者使用药品的时机,或
6.基于儿科人群的安全考虑,宜先观察成人研究的安全性或有效性,或
7. 其他合理的延缓理由。
FDA给药品研发企业提供了一份申请延缓清单,以帮助其为申请延缓搜集足够的信息。为了获准延缓,申请人必须提交延缓评估的理由及证明资料、计划或进行中的研究综述、研究正在进行或将会尽早进行的证据。
2015年FDA批准的27个NASs中,获准延缓的有14个。与豁免类似,延缓也可以针对特定年龄段的儿科人群。因此,一个产品可针对特定不同年龄段的儿科人群同时获准部分豁免和延缓。
PREA可要求进行不同类型的儿科研究。FDA会考量证明每个产品安全性有效性需搜集的数据类型。若可能有毒性,FDA可能会要求申请人先在幼龄动物中进行临床前毒理学研究,再进行临床试验。若批准的成人用处方不适用于儿科人群,FDA可能会要求对儿科人群适用的处方进行分析研究。有些情况下,为了避免用儿童进行不必要的重复试验,也可用已有数据进行推论。
表2列出了本文分析的产品被要求进行的研究类型。尽管只包括了2015年批准的NASs,对于一般被要求进行的研究,本表仍很具代表性。要求的试验通常都是出于PREA有效性和安全性的顾虑。本文涉及产品的申办方,甚至100%都被要求提供安全性和有效性的评估。
经常要求提供的还有药代动力学和药效学数据,2015年批准的NASs中86%的儿科研究计划都包括了药代动力学和药效学试验。一半被要求进行耐受性研究。14个儿科研究计划中,只有3个(21%)包括了临床前毒理学研究。
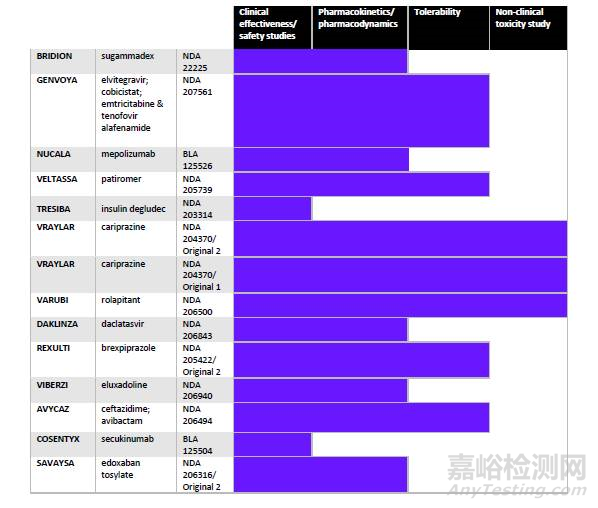
表2 PREA要求进行的研究类型
来源:科睿唯安Cortellis 药政法规数据库:美国法规情报报告--儿科研究和发展综述
最后,本文分析了2015年最常进行儿科研究的治疗领域,以及与之相反获准全部豁免的治疗领域。意料之中,获得全部豁免的主要为成人适应症的治疗领域。因此,近50%的全部豁免为用于治疗成人癌症和心血管疾病的产品(见图3)。
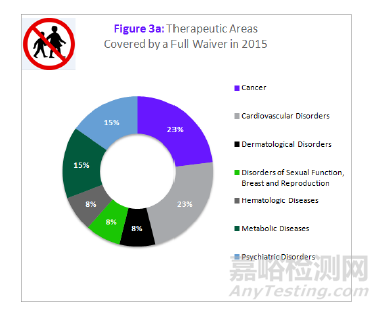
来源:科睿唯安Cortellis 药政法规数据库:美国法规情报报告--儿科研究和发展综述
针对此问题,FDA在“PREA申请指南”中给出了有可能因适用于特异性疾病而豁免的成人适应症,包括:年龄相关性黄斑退行性改变,阿尔茨海默症,肌萎缩、动脉硬化、不孕不育、更年期症状、骨关节炎、帕金森综合症、基底细胞癌和鳞状细胞癌,乳腺癌,结肠直肠癌,子宫内膜癌,毛细胞癌、肺癌、口咽癌、卵巢癌、胰腺癌、前列腺癌、肾细胞癌和子宫癌。
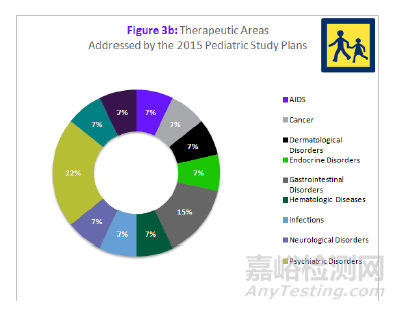
来源:科睿唯安Cortellis 药政法规数据库:美国法规情报报告--儿科研究和发展综述
图3b列出了2015年儿科研究计划涉及的多种治疗领域,治疗领域超过10个。其中占比最大的为精神疾病(22%)和血液疾病(15%)。今年来儿科研究涉及的治疗领域并没有太大变化。2012年FDASIA签署前就对其进行了统计,但签署后这一数据并没有太大变化。实际上,过去5年来,儿科研究涉及的治疗领域并无重大进展。将2008-2014年间的儿科研究涉及的治疗领域与2015年的进行比较,会发现多年前就是图中的治疗领域。当然,在过去的4年里,开展儿科研究的数量有相当大的增幅。
过去十年中,越来越多的意识到儿童对药物的反应不同于成人。这对促进不断研发适应儿童生长发育特点、年龄和治疗反应的药物有积极的影响。使得成人药品超说明书用于儿童的现象大量减少,开展儿科研究的数量增加。PREA和BPCA的实施无疑是取得如此进展的原因之一。在PREA和BPCA成为永久性法律之前,超过80%的成人药品超说明书用于儿童,其安全性和有效性毫无保障。而在这之后,此数据降至低于50%。很明显FDA的胡萝卜加大棒策略取得了成功。
FDA强烈建议申请人研发新药的过程中尽早与FDA相关负责部门会面。最好在II期临床结束后III期临床开始前,向FDA提交儿科研究计划。在研发项目的早期确定儿科研究要求有助于申请人和FDA间更好的协作,会允许申请人提交满足PREA和BPCA的必要文件。
尽管为鼓励进行儿科临床试验做出了很大努力,仍然存在保护这一弱势群体,使其免受不必要和重复试验的担忧。监管机构,尤其是FDA和EMA之间的合作和信息共享有所加强。政府也在研究更为协调的儿科药政法规要求。协调保护儿童免受试验药物和继续推进儿科研究这两者的平衡非常重要。
对药品申请人而言,研发儿科产品仍有挑战。获得6个月市场独占期的奖励虽然很有吸引力,但儿科试验通常都是大规模且花费巨大的。实际上,BPCA的激励并不能确保投资回报率。此外,这一“胡萝卜”对产品已经没有专利保护的申请人来说并无吸引力。对这些过了专利期的药品而言,BPCA鼓励申请人与美国国立卫生研究院(National InstitutesofHealth,NIH)沟通,NIH有针对儿科研究的研究基金。
FDASIA签署5年后,FDA可以对其PREA和BPCA的策略做一个总结了。总体感觉对儿科患者及其家属、药企和卫生部门有积极影响,但FDA可能想要对结果进行更深入的研究。另外一些方面还存在顾虑:BPCA的激励对药品研发者而言是否仍然具有优势?毕竟儿科临床试验的花费相当高。如何激励过了专利期的药品?能在与儿科适应症有关但尚未在儿童中进行研究的治疗领域有所进展吗?如何鼓励申请人尽早与审评人会面,以确保形成高质量的儿科研究计划?通过PREA不符合信,FDA可以行使其对不符合PREA的申请人的权利吗?

来源:科睿唯安


