在2022年11月,由RDPAC 的药学团队对中美欧药学技术指导原则和指南进行了调研与对比,总结分析了中美欧指南的标准差异及实施情况差异,采用了主题词及分类方式、深度对比、报告撰写和定稿流程,最后呈现研究报告,给到药审中心参考。
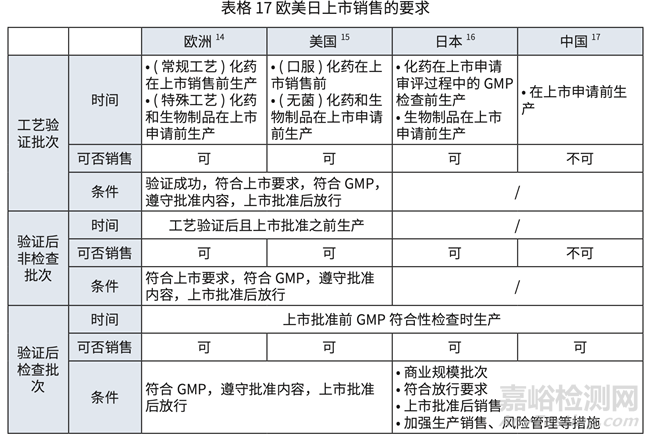
目前,ICH 主要国家实践上,允许批准前生产批次(包括验证批次,检查和非检查批次) 上市销售。而我国 NDA 和上市后变更中的批准(和备案)后执行日期定义不清晰 , 造成批准(或 备案)前生产批次可否上市不清晰,给患者用药和供应链造成一定风险。建议以问与答的形式, 明确对执行日期的定义,明确在上市许可批准和变更批准或备案前生产的批次满足一定条件后, 比如 GMP 条件,放行条件等,即可上市销售。推荐加急制定。
原因分析:
• 欧美日现行指南:
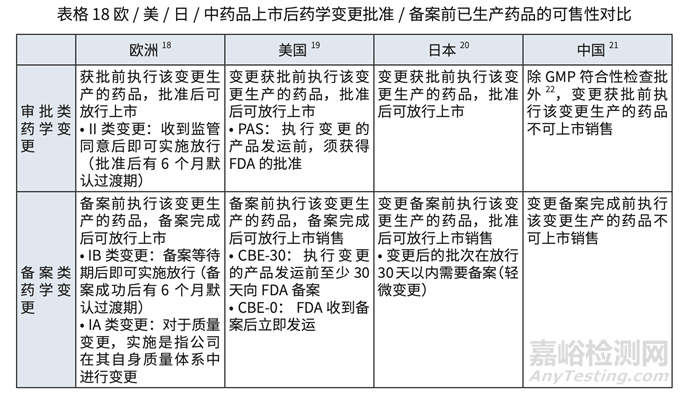
对于欧美日中,批准前生产批次(包括验证批次,检查和非检查批次)可上市销售的要求 对比详见表格 17 和表格 18.
• 中国实施问题
NDA 和上市后变更中的批准(和备案)后执行日期理解为生产日期 , 造成批准(或备案) 后方可生产,之前生产的批次无法上市。
以“生产日期”作为注册批准(药品上市许可和上市后变更)实施监管节点在实际工作中 的影响,RDPAC 在会员公司内发起了调研(报告请参见附件),从获得的数据分析发现:
1. 新药供应中国市场的延迟:药品上市许可获批后在执行“批准后方可生产”的情况下, 境外生产新药(临床急需品种除外)从上市批准到供应中国市场的时间差约为 7 个月(中 位值),且年度生产批次少的罕见病新药通常需要更长时间,导致患者难以尽早获益已 批准的新药。对于境外生产的临床急需品种,由于允许上市供应批准前生产的批次,时 间差可缩短为 2 ~ 3 个月,与境内生产的药品从批准到供应的时间差相当。
2. 增加供应短缺和患者可及性的风险:药品上市后药学变更因境内外不能同步报批且需执 行“批准后方可生产”的要求,从境外实施变更到境内供应变更后药品之间的时间差长 达 19 个月(中位值)。其中,执行“批准后方可生产”引起的时间差约 9 个月(中位值)。 由此,该期间行业仅能依靠提前积压数千万至数亿元资金备货以维持中国市场的供应, 且备货通常仅可维持最多 9 个月的供应,发生断药的风险较高。
总之,以“生产日期”作为注册批准实施监管节点对确保临床患者用药可及性以及行业运营带来很大的困难。
• 相关的国内监管法规 :
《中华人民共和国药品管理法实施条例(修订草案征求意见稿)》第六十条【注册前规模批药品上市销售】质量标准、生产工艺与注册证书一致的商业规模批次药品,其生产过程符合药品生产质量管理规范的,在取得药品注册证书后,符合产品放行要求的,可以上市销售;药 品上市许可持有人应当对其加强生产销售管理和风险管理。
建议:
尽快出台 Q&A 对执行日期进行明确定义,明确在上市许可批准和变更批准或备案前生产 的批次满足一定条件后,比如 GMP 条件,放行条件等,即可上市销售。
1. 充分落实 MAH 制度,按照《药品管理法》“第二十四条”精神,“以药品的‘质量受权 人放行日期’(简称放行日期)作为注册批准后的实施时间,适用于药品上市许可和上 市后变更的注册批准。”
2. 境外药品无论在进口环节还是在市场流通环节,注册批准的执行情况均可通过查验随货 同行的药品检验报告书(CoA)上载明的“放行日期”,并通过相关文件溯源以达到有效监管的目的。
参考文献
1.国内外药品技术指导原则体系对比研究 (药学部分》,国家药品监督管理局药品审评中心,中国药品监督管理研究会,药品监管研究国际交流专业委员会,中国外商投资企业协会药品研制和开发工作委员会(RDPAC),2022年11月



