您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2025-09-06 18:34
摘 要 / Abstract
为进一步加强二代基因测序相关体外诊断试剂类产品监督管理,推动产业高质量发展,国家药品监督管理局组织制定了《二代基因测序相关体外诊断试剂分类界定指导原则》。该原则是二代基因测序相关体外诊断试剂制造商和相关监管人员开展工作的重要依据。本文介绍了该原则的制定背景,并对重点内容进行了解读和探讨,旨在帮助相关人员准确理解,进而统一我国二代基因测序相关体外诊断试剂的分类管理标准。
关 键 词 / Key words
体外诊断试剂;二代基因测序;分类界定;指导原则;分类管理
2025 年6 月, 国家药品监督管理局(以下简称国家药监局)发布《 二代基因测序相关体外诊断试剂分类界定指导原则》(以下简称《指导原则》)[1],旨在应对二代基因测序(next-generation sequencing,NGS)技术快速发展带来的监管适配性问题,推动产业规范化发展。该文件与《体外诊断试剂分类目录》及配套通告[2-4]形成协同监管体系,标志着我国针对NGS 相关体外诊断试剂的监管工作迈入“包容创新、风险精细化管控” 新阶段。《指导原则》的核心要点在于统一分类标准,明确提出以技术功能分类法替代传统的产品形态分类法,将NGS 相关体外诊断试剂根据核心功能单元拆分为核酸提取纯化、文库构建、测序反应3 个模块,并确立以靶标识别能力作为类别判定的核心依据。本文将围绕《指导原则》的制定背景及主要内容展开介绍和解析。
01《 指导原则》制定背景
NGS 技术发展迅速,其相关体外诊断试剂涉及核酸提取纯化、文库构建及测序反应等环节。其中,文库构建与测序反应环节所用试剂组分繁多,这给监管工作带来了不小的挑战。在实际监管过程中发现,部分企业将本应归为第三类体外诊断试剂的文库构建试剂(含靶向探针)以第一类测序反应通用试剂名义进行备案,该做法模糊了产品的风险等级。为进一步加强NGS 相关体外诊断试剂的监督管理,推动行业高质量发展,国家药监局特制定《指导原则》。该原则在参考国内外相关分类文件、依据《体外诊断试剂分类规则》[5-6] 确立分类框架的基础上,针对监管过程中存在的疑点和难点问题,通过文献检索、咨询调研及专家研讨等方式予以厘清和解决。
02NGS 技术与相关体外诊断试剂分类管理探讨
2.1 NGS 技术
NGS 技术是继 Sanger 测序技术后的一项革命性生物技术,能够同时对数百万甚至数十亿个脱氧核糖核酸(deoxyribonucleic acid,DNA)片段进行测序,实现了大规模、高通量测序的目标。相较于其他测序技术,NGS 技术在速度、通量和价格方面均具有优势,而且可以同时对多个基因区域、多种类型基因变异进行识别和检测。凭借这些优势,NGS技术在分子诊断、医药健康等多个领域展现出广阔的应用前景[7-9]。
2.1.1 NGS 技术检测流程概述
目前,国内外主流的NGS 技术,其核心检测原理具有高度相似性(图1)[7],主要检测流程包括核酸提取纯化、片段化与文库制备以及测序。其中,核酸提取纯化过程相对独立且容易理解,因此本文将重点阐述其他关键步骤。
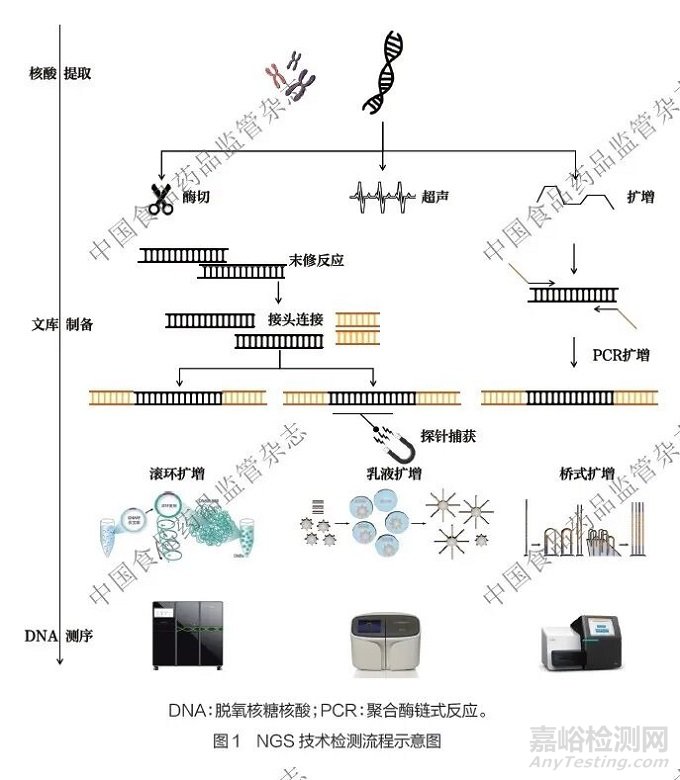
片段化与文库制备。该步骤首先将待测的长链核酸,通过物理或酶学方法打断为特定长度的短片段,这些短片段的长度范围需严格匹配测序平台的兼容性要求。随后进行文库制备, 即在片段化产物两端连接必要的功能序列, 包括用于样本多重识别与溯源的样本标签(Index或Barcode)、测序引物结合位点(Adapter) 等, 采用的试验步骤主要有末端修复、加A尾、加接头等,形成包含基因组信息的初始文库。若检测目标为特定基因区域, 则需在此阶段应用靶向富集技术, 如探针杂交捕获或目标区域多重聚合酶链式反应(polymerase chain reaction,PCR) 扩增, 从初始文库中富集目的片段,从而得到以检测目的片段为主的样本文库。
成簇和测序。在开展测序工作之前,需进行核酸提取(基因组DNA), 以酶切、超声打断或多重扩增等方式构建文库,以添加接头或靶向捕获等方式通过PCR 技术(如滚环扩增、乳液扩增、桥式扩增等)对文库中的单个DNA 片段进行克隆式扩增。该步骤利用测序试剂盒提供的酶与底物,在固相载体(如测序载片或芯片)上形成高密度、包含大量相同拷贝的片段簇(Cluster)。片段簇可直接在测序芯片上生成,也可以另行制备后再加载至芯片上,最终构成高密度DNA 阵列,为后续进行高灵敏度的信号检测奠定基础。测序过程是指对高密度DNA 阵列执行循环生化反应与信号采集。该过程采用循环机制:首先进行酶促合成反应(依赖聚合酶或连接酶催化碱基特异性延伸),随后执行信号捕获(光学平台采集荧光/ 化学发光图像,半导体平台实时监测氢离子浓度变化),经多轮循环后,生成原始测序序列[10]。
2.1.2 NGS 相关体外诊断试剂
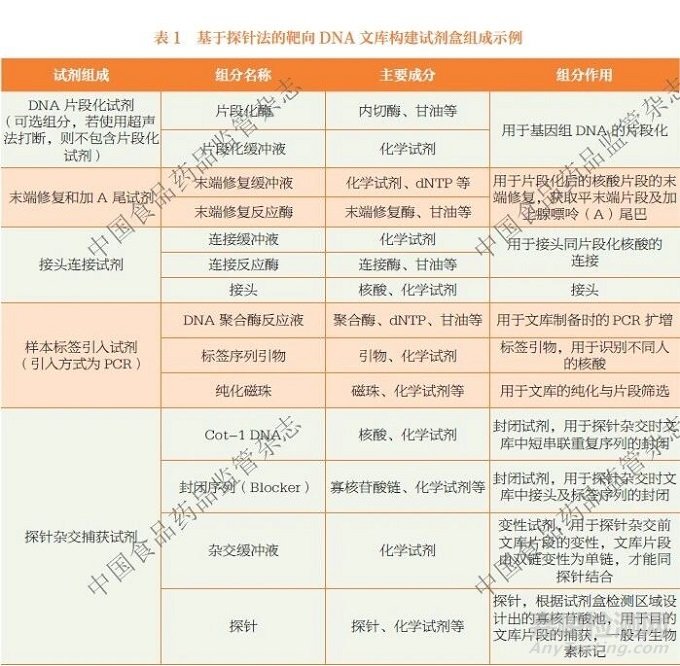
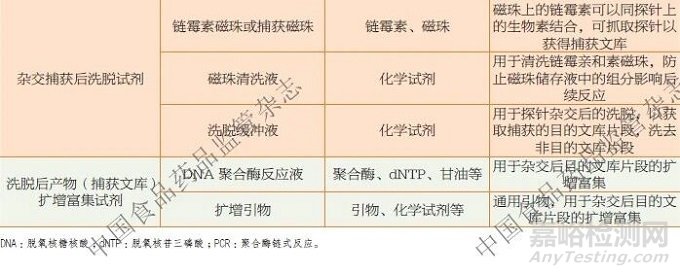
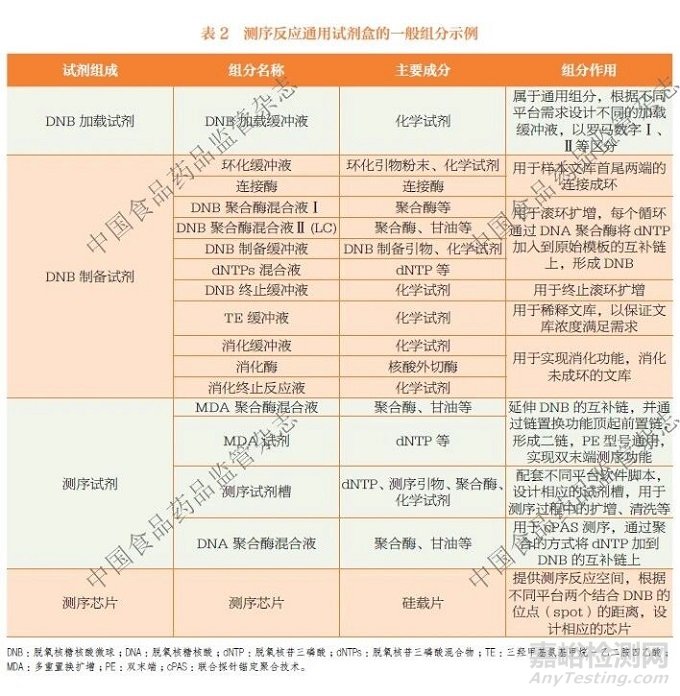
从技术层面上看,NGS 相关体外诊断试剂按核心功能单元,可以拆分为核酸提取纯化、文库构建、测序反应3 个模块。虽然文库构建试剂和测序反应通用试剂都有PCR 过程,且一般都包含引物、缓冲液、脱氧核苷三磷酸(deoxyribonucleoside triphosphate,dNTP)、DNA聚合酶、连接酶以及化学试剂等成分,但由于二者的主要作用不同,以及各自含有特有的组成成分或部件,因此是可明显区分的(表1、表2)。
2.2 文库构建试剂分类界定要点
文库构建试剂的主要作用是进行文库制备,其组成成分通常涵盖文库构建过程中特有的片段化酶、片段化缓冲液、接头、单/ 双标签序列引物(Index 或Barcode)、一/ 二轮扩增反应液、探针(或目标序列特异性引物)、捕获磁珠或链霉亲和素磁珠、洗脱反应液或洗脱缓冲液等。如果涉及核糖核酸(ribonucleicacid,RNA)建库,建库试剂盒还会包含逆转录酶或逆转录反应液。文库构建试剂所包含的特异性引物或探针等关键组分,直接决定了检测的准确性、特异性等核心性能指标,且相关试剂具有明确的临床诊断性能,是检测风险的主要来源,因此明确按照第三类体外诊断试剂对其进行管理。
《指导原则》强调,文库构建试剂应能实现完整的文库构建功能;同时,明确禁止将同一建库流程中的试剂拆分为多个独立试剂盒进行申报(如单独申报仅包含靶向捕获功能的试剂盒)。这一规定旨在确保该类高风险试剂管理的完整性和有效性。
2.3 测序反应通用试剂分类界定要点
测序反应通用试剂须在特定的NGS 平台上使用,其主要功能是实现测序过程中的信号采集,最终获取文库样本的序列信息。
在国际监管层面,测序反应通用试剂, 在美国由食品药品监督管理局(Food and Drug Administration,FDA) 按照Class I 进行管理;在欧盟, 依据《体外诊断医疗器械法规》(In Vitro Diagnostic Medical Devices Regulation,IVDR), 按照Class A 进行管理;在俄罗斯、加拿大、韩国、马来西亚等其他主要国际市场,也均被划定为低风险类别进行监管。此类试剂的核心特性是平台通用性和非检测靶标特异性:一方面,仅适用于特定测序平台所有类型的测序文库;另一方面,仅包含测序过程必需的通用组分(涵盖测序前必要预处理环节的试剂组分),但禁止含有任何靶标特异性序列或文库构建组分(如样本接头等)。这类试剂风险相对较低,因此按照第一类体外诊断试剂进行管理。
2.4 注册单元要点解析
测序反应通用试剂与文库构建试剂配合使用方可完成测序功能,鼓励二者组成同一注册单元,共同申报第三类体外诊断试剂注册。随着技术不断迭代升级,如当前已有仪器将文库构建与测序功能整合于一体,预计此类集成化设计的应用将愈发普遍。
为了更具普适性和灵活性,《指导原则》允许测序反应通用试剂与文库构建试剂作为不同单元分别进行申报,要求申请人应当从实现功能、技术特征、结构组成等角度,明确二者间的划分。文库构建试剂注册时应当明确适配的测序反应通用试剂。测序反应通用试剂备案时应当明确适配的仪器品牌、型号(需适配已取得医疗器械注册证的NGS 仪器)。
03《 指导原则》出台对科学监管的意义
通常情况下,文库构建试剂与测序反应通用试剂在技术和专利归属等方面存在差异。文库构建试剂由相关制造商基于检测方向独立研发;而测序反应通用试剂的开发与测序仪的硬件结构、液路系统、成像系统等高度适配,主要由测序平台制造商负责开发和改进。《指导原则》的出台,对于维护测序行业技术开发生态、推动测序技术创新发展具有重要意义,具体体现在以下3 个方面。
(1)采用技术功能分类法替代传统的产品形态分类法,实现风险精细化管控和科学监管政策延续。《指导原则》明确将测序反应通用试剂作为独立产品,按第一类体外诊断试剂进行管理,而非强制将其与第三类体外诊断试剂绑定。这一规定有效避免了强制合并注册单元可能带来的系统性风险,确保了合规的第三类文库构建试剂注册证依然有效,显著降低了产业链重构风险,有力保障了合规企业生产经营的连续性。
(2)分类边界精细化,包容创新,实现管理类别界定清晰化与标准化。《指导原则》厘清了原本模糊的技术边界,明确了文库构建试剂(第三类)与测序反应通用试剂(第一类)的功能区分,避免文库构建试剂等组分混入测序反应通用试剂,精准打击将第三类文库构建试剂(含靶向探针)伪装为第一类测序反应通用试剂等“高类低备”的乱象。
(3)平衡监管刚性与发展弹性,引导产业高质量发展。《指导原则》为测序反应通用试剂增加了3 个入市门槛:一是要求配套使用的仪器需先获批医疗器械注册证;二是明确限定配套的适用机型范围;三是规定测序反应通用试剂不得含有文库构建功能,也不能与文库构建试剂穿插使用。此外,《指导原则》通过规范测序反应通用试剂的备案要求,增加“限定条件”,降低了临床滥用风险,遏制了市场失序行为。这一系列规定既保障了“提取- 建库-测序”整体检测流程的顺畅进行,又满足了临床实际需求,有力引导了产业的高质量发展。
04结 语
《指导原则》通过科学合理的设计,进一步强化了风险管控力度,有效堵住了部分企业试图以“高类低备”规避临床确认与技术审评的监管漏洞。其采用国际通行的独立注册单元管理模式,实现了我国医疗器械监管与国际监管标准的顺利接轨。在实践中,《指导原则》有利于平衡监管刚性与发展弹性,为分类存疑的医疗器械分类管理提供了范式参考。同时,《指导原则》引导产业进行技术创新、技术竞争、技术出海,有力驱动了产业向高质量发展转型,进而推动NGS 技术更广泛地应用于临床,造福更多患者。

来源:Internet


