口服溶液剂具有给药剂量灵活、患者顺应性好等优点,尤适于吞咽困难的老年患者和儿童人群,是儿童常用剂型。近年来,国家药品监督管理局(NMPA)、美国食品药品监督管理局(FDA)、欧洲药品管理局(EMA)发布了多个化学药品儿童用药相关指导原则。本研究重点对上述指导原则进行梳理,对儿童用口服溶液审评中原辅料选择、质量控制等问题进行总结,以期为儿童用口服溶液的研究和申报提供参考。
一 原辅料方面
1.1 原料药
在新药开发早期,建议综合考虑目标年龄段儿童的情况、剂型、原料药关键理化特性(生物药剂学分类系统分类)和稳定性等因素,选择适合的原料药形式(游离酸/碱、盐、晶型、溶剂化物等)及粒径等属性,以提高药物的可接受性[1]。对于口服溶液剂,原料药可选择具有更高溶解性的盐型,但需关注药物体内药动学特征的改变是否会影响药品的安全性和有效性[2]。
1.2 辅料
儿童用口服溶液剂的辅料通常包括着色剂、矫味剂(如甜味剂)、抑菌剂、增溶剂[1],主要作用是改善药物溶解度、提高药物稳定性、改良适口性等。
对于儿童用口服溶液剂的仿制药,辅料种类和用量建议与原研产品保持一致,申请人可提交与参比制剂中抑菌剂、矫味剂等不同的处方,但需阐述选择的理由,对于可能影响药物吸收或生物利用度的辅料需谨慎考量。对于儿童用口服溶液剂新药,除按照相关技术要求考察原辅料相容性外,需重点关注辅料在拟定儿童人群中的安全性。《儿童用药(化学药品)药学开发指导原则(试行)》指出[1],在降低风险以及确保产品的功效、稳定性、适口性、微生物控制和剂量均匀性的前提下,应尽可能使用最少种类和最低用量的辅料;应关注潜在致敏或过敏性的辅料;注意评估辅料对原料药的吸收和生物利用度的潜在影响。
在辅料种类和用量方面,应考虑是否有相同给药途径下的儿童药品的使用、给药周期及服用剂量是否相当。如引用新的辅料,应提供辅料或制剂的临床前安全性试验资料。
对于含有抑菌剂的制剂,应照《中华人民共和国药典》2020 年版(ChP 2020)四部通则 1121 进行抑菌效力试验,在稳定性试验末期进行抑菌效力检查。部分抑菌剂,例如苯甲酸及其盐类可能增加新生儿(4 周以下)发生黄疸的风险,对羟基苯甲酸酯可能引起过敏反应,因此引入时需关注抑菌剂的潜在风险。
辅料的特殊安全性问题尤其需要关注。例如,丙二醇可能在 4 岁以下儿童体内蓄积,可能产生的不良反应包括中枢神经系统毒性、高渗透压引起的腹泻等;乙醇有引起儿童患者急性或慢性中毒的风险[1]。香精是常用的矫味剂,其成分包括酯类、醛类(如柠檬醛、香草醛、苯甲醛、癸醛、乙醛)等,其中小分子醛类为致突变杂质,针对其在儿童人群中的使用需严格检查和控制;对于单次服用剂量大的儿童口服溶液,致突变杂质的控制尤为重要。在申报资料中建议明确提供小分子醛类的种类,建立灵敏度符合要求的分析方法对致突变杂质进行检查,根据人用药品技术要求国际协调理事会(ICH)发布的《M7:评估和控制药物中 DNA反应性(致突变)杂质以限制潜在致癌风险》等指导原则进行评估,建立合理的控制策略。
除 NMPA 发布的指导原则外,在儿童口服溶液研发过程中还建议研究者关注国外监管机构和行业协会发布的相关要求和信息。欧洲儿童用药行业协(European Paediatric Formulation Initiative,EuPFI)组织创建了儿科辅料安全性和毒性(Safety & Toxicity of Excipients for Paediatrics,STEP)数据库,收集了欧美药品监管机构批准上市的药品中部分常用辅料在儿童人群中的用量,对辅料种类、用量的选择提供了安全性参考信息。
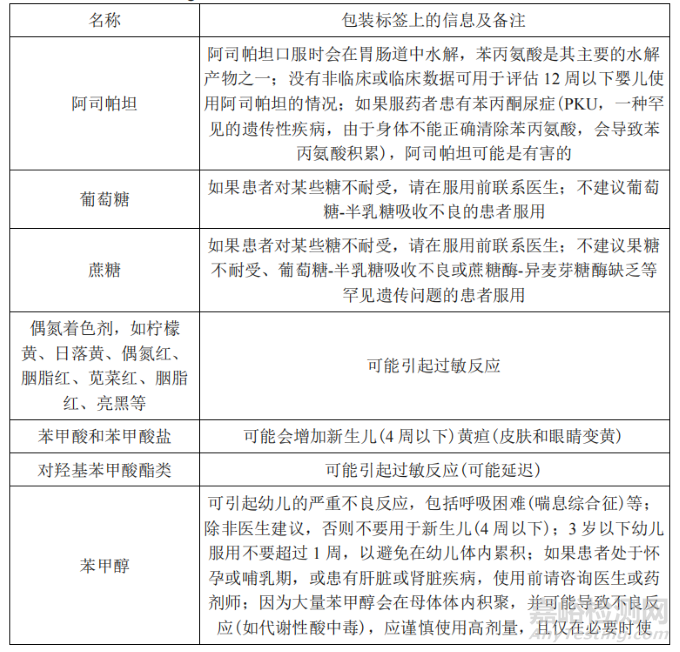
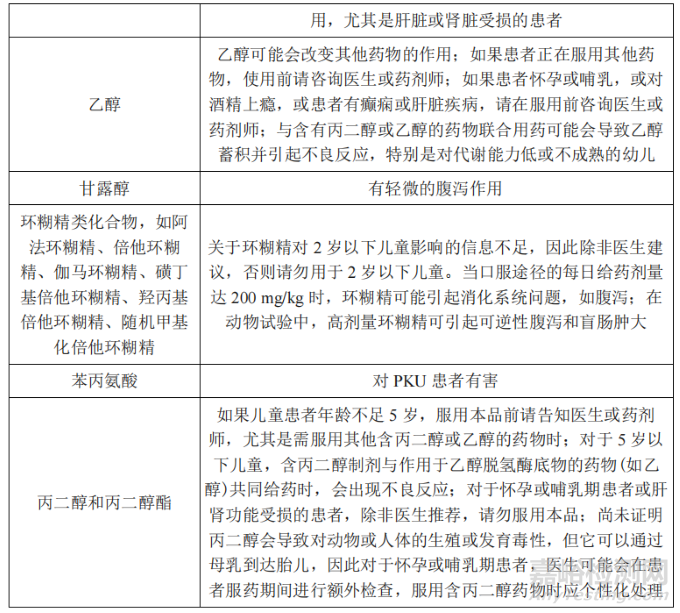
此外,EMA 在《人用药品说明书和包装标签中辅料相关信息指导原则》的附件中提示部分辅料具有潜在风险[3],建议研究者结合患者生理特点、给药剂量、治疗周期、辅料作用等方面因素综合考量。部分口服给药途径制剂中需关注的辅料信息如表 1 所示。
表 1《人用药品说明书和包装标签中辅料相关信息指导原则》收载的部分辅料信息
Tab.1 Information on Some Excipients Included in《Excipients in the Labelling and Package Leaflet of Medicinal Products for Human Use》
二 质量控制方面
2.1 微生物控制
口服溶液剂在生产和使用过程中易受到污染,若处方中含糖类等甜味剂,微生物易滋生。美国 Lannett 公司于 2019 年召回了其产品左乙拉西坦口服溶液,原因为枯草芽孢杆菌污染[4];FDA 在普萘洛尔口服溶液、马来酸依那普利口服溶液的审评报告中均要求对洋葱伯克霍尔德菌(Burkholderia cepacia)进行检测并建立控制策略。
药品研发质量管理体系控制要点以及在委托研发、注册申报中的应用
枯草杆菌(Bacillus subtilis)群危害健康的可能性取决于微生物污染程度、治疗剂量和持续时间、患者的潜在状况等因素,免疫功能低下的患者可能会发生严重感染。洋葱伯克霍尔德菌广泛存在于土壤、植物和人体内,能在人与人之间进行传播,在免疫功能低下患者(如囊性纤维化和慢性肉芽肿病患者)中易引起感染,而且耐多种化学抑菌剂[5]。调研发现,药品生产中使用的纯化水、治疗门诊样品中均检出洋葱伯克霍尔德菌,且其为检出量最高的污染菌。2017 年 5 月 FDA 发布声明告知药物制造商,洋葱伯克霍尔德菌复合物在非无菌水性基质制剂中存在污染风险[6]。2021 年 9 月 FDA 发布了《非无菌药品生产中的微生物质量考量》行业指南草案[7],阐述了非无菌水性基质药品受洋葱伯克霍尔德菌和其他微生物污染导致的药品不良事件、风险及控制。美国药典(USP 2022)<60~62>章阐述了洋葱伯克霍尔德菌复合物试验、微生物计数试验、特定微生物测试等内容。
药品审评中心(CDE)在《化学药品创新药上市申请前会议药学共性问题及相关技术要求》《化学药品创新药Ⅲ期临床试验前会议药学共性问题及相关技术要求(征求意见稿)》 中均要求参照相关技术要求对洋葱伯克霍尔德菌进行研究,制定相应的控制策略[8—9]。对于儿童用口服溶液,除要按照 ChP 2020 四部附录进行微生物限度检查和控制外,还需对洋葱伯克霍尔德菌进行检查和控制。
ChP 2020 四部通则 1107《非无菌药物微生物限度标准》中明确要求根据原辅料及其制剂的特性和用途、制剂生产工艺等因素,对其他可能存在的具有潜在危害的微生物进行检查。申请人可针对制药用水等引入洋葱伯克霍尔德菌的风险进行评估,列入放行质量标准或注册标准。
口服溶液属于非无菌制剂,对于多剂量口服溶液需模拟临床实际使用情况,考察使用中的稳定性。考察项目包括物理化学稳定性、微生物限度等。研究样品建议包括稳定性试验末期样品。根据研究结果,在说明书相应项下列出使用中的稳定性信息,指导临床合理用药。
2.2 给药器及给药准确性
口服溶液的给药量需精准控制。例如某注册申请拟定药品说明书中口服液用量为每天 0.8 ml,但附带提供的量器最低刻度为 1.5 ml,无法满足临床准确给药的需要。FDA 在 2011 年发布了《OTC 液体口服制剂分剂量量具指导原则》[10],指出液体口服制剂的给药装置应标明经校准过的液体测量单位。
建议结合儿童口服溶液特性(单次最小给药体积、黏度等),选择精密度符合要求的量取装置,采用色谱法测定药物量等方法对附带给药器的准确性进行考察,以确认给药的准确性。
2023年eCTD相关法规与递交实操研讨班
同时,对于儿童口服溶液,需注意参照相关技术要求考察给药器和药液的相容性。
部分儿童口服溶液原研产品中附带的给药器是聚丙烯材质的精密刻度注射器,以防止幼儿口服时咬合造成给药器破碎,引发风险。仿制产品若附带的给药器采用玻璃注射材质,因幼儿口服给药时咬合造成给药器破碎引发的风险将显著增加,不建议使用。
三 总结与展望
我国近年先后出台了一系列政策文件,以鼓励和促进儿童药物的研发生产。新的《药品管理法》《药品注册管理办法》明确提出鼓励儿童用药品的研制和创新,对儿童用药品予以优先审评审批。口服溶液是常用剂型,建议研究人员参照相关行业指南及指导原则,关注辅料安全性风险、质量控制等方面,建立相关控制策略,提升儿童用药质量。