您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-08-11 19:43
一、如何申请分类界定?
答:对新研制的尚未列入《医疗器械分类目录》或分类界定文件的产品即未在我国境内上市的全新产品;或者与已上市产品相比,产品的技术原理、结构组成、使用部位或技术特点、预期目的等发生了影响产品分类的实质性变化,依据《医疗器械分类目录》或分类界定文件难以确定管理类别的产品,注册申请人可以依据《医疗器械分类规则》初步判断产品类别并按照国家药品监督管理局要求(网址: https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjylqx/20170926173301389.html),登陆“医疗器械分类界定信息系统”申请分类界定。
分类界定工作流程如下图所示。
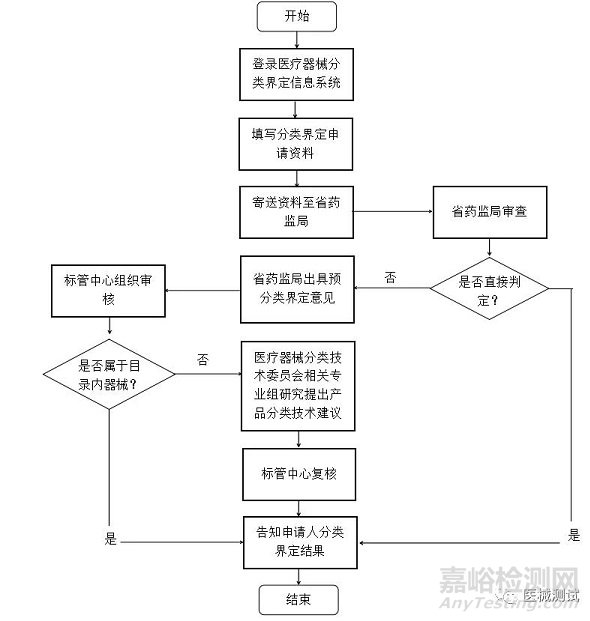
二、分类界定时限要求?
答:依据《医疗器械监督管理条例》(国务院令第739号)第二十三条,申请类别确认的,国务院药品监督管理部门应当自受理申请之日起20个工作日内对该医疗器械的类别进行判定并告知申请人。
三、新研制的尚未列入分类目录的医疗器械,是否一定要申请分类界定?
答:不一定,可以依据第三类医疗器械产品注册的规定直接向国家局申请产品注册,也可以依据分类判定规则判断产品类别并按国家局要求申请分类界定。
四、注册申报产品含有多种规格型号,注册检验报告有何要求?
根据国家医疗器械相关法规并结合本省实际,注册申请人可提供以下形式之一的检验报告:
1、所有申报型号规格的全性能检验报告。
2、有资质的检验机构出具的典型型号的全性能检验报告及产品型号覆盖评价报告。
3、有资质的检验机构出具的典型型号全性能检验报告、医疗器械注册申请人提供的典型性说明、典型型号与被覆盖型号的具体差异性分析,必要时提供检验机构出具的差异项目检验报告,推荐提供被覆盖型号的自检报告或者第三方委托检验报告作为佐证。
注:提供自检报告的需符合《医疗器械监督管理条例》(中华人民共和国国务院令 第739号)及后续相关配套法规文件的要求。

来源:Internet


