您当前的位置:检测资讯 > 生产品管
嘉峪检测网 2019-09-06 09:21
ISO14971是国际公认的用于医疗器械风险管理的标准,目前国内遵循的标准YY/T0316-2016就是等同转化的ISO14971-2007修正版。标准中提到,在医疗器械投入生产时,风险管理的工作依然没有停止,制造商要从生产和生产后的活动中,取得可能影响风险管理决策的数据和信息。
下图是YY/Y0316-2016中的风险管理流程图,图中的流程显示生产和生产后的活动是属于风险管理的过程。
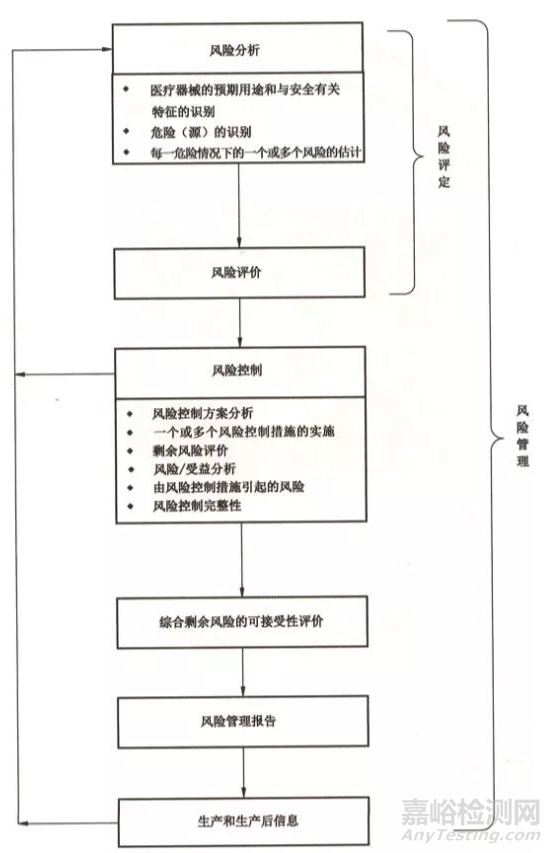
既然标准有明确要求风险管理需要包含生产和生产后的活动,那具体应该怎么做呢?YY/T0316-2016进行了简单的描述,更具体的要求我们可以参考今年发行的的ISO FDIS 14971-2019,该版本已经结束了征求意见的阶段,相信不久便会发行正式版本。新版的ISO14971对生产和生产后的活动内容规定得更为具体,主要分为三个步骤:
搜集信息
审查信息
采取行动
我们就来看看这三个步骤具体包含了哪些要求:
1. 搜集信息
首先,制造商需要搜集生产和生产后的信息,这些信息主要包括:
生产过程和生产过程的监视中产生的信息,比如:生产中不合格品的趋势分析的信息
用户生成的信息,比如:用户的反馈和投诉
负责安装,使用和维护医疗设备的人员所产生的信息,比如:需要安装、调试、维护的医疗器械,通常需要专业的技术人员上门服务,由他们在现场安装和服务过程中产生的信息
供应链产生的信息,比如:经销商和进口商的反馈信息
公开的信息,比如:不良事件的通告和召回的信息
与公认的现有技术有关的信息,包括最新的标准,公开发布的医疗器械的应用数据,替代医疗器械或治疗的有效性等等。
类似医疗器械的公开信息,包括不良事件通报的信息、召回的信息、公开的临床文献中的信息、来自数据库中的信息等等。
2. 审查信息
制造商将信息搜集过来后,接下来要做的是审查这些信息,筛选出那些与器械安全相关的信息,特别要注意以下这几种情况:
存在以前未识别的危险或危险情况;
危险情况引起的风险估计不再能接受;
与预期用途的收益对比,总体的剩余风险不再能接受
公认的现有技术发生了变化
制造商要将信息的审查结果记录在风险管理文件中,用于下一步风险管理文件合规性的检查。
3. 采取行动
最后,制造商就要针对审查出来的这些与医疗器械安全有关的信息,采取进一步行动。采取的行动主要针对两方面:一方面是要针对器械本身,另一方面是针对风险管理过程。
1)关于医疗器械
制造商需要审查风险管理文件,确定是否有必要重新评估风险,或是评估新风险。
如果剩余风险不再可接受,则要评估对先前实施的风险控制措施的影响,并且需要之前的风险控制措施作为修改医疗器械的输入;
制造商要考虑对市场上的医疗器械是否需要采取必要的行动
所有的决定和行动都要记录在风险管理文件中。
2)关于风险管理过程
制造商要评估这些信息对之前实施的风险管理活动的影响;
此评估结果将作为最高管理层评审风险管理过程适宜性的输入。
结语
今天分享的主要内容就到这里,熟悉医疗器械法规的朋友可能对于今天的内容并不陌生,各国的医疗器械法规关于器械上市后监督也要求关注器械的风险相关的内容,所以在实际操作时,大家可以将两者结合起来做,定能事半功倍。

来源:启升资讯


