您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-07-22 18:41
近日,国家药品监督管理局批准,现予发布《心可宁胶囊中酸性红73检查项补充检验方法》等17项药品补充检验方法,内容如下。
1.心可宁胶囊中酸性红73检查项补充检验方法(BJY201901)
2.妇科止带片中金胺O检查项补充检验方法(BJY201902)
3.心宁片中赤芍、三七茎叶植物组织检查项补充检验方法(BJY201903)
4.银柴颗粒中灰毡毛忍冬皂苷乙检查项补充检验方法(BJY201904)
5.心可舒胶囊中人参皂苷Rb3检查项补充检验方法(BJY201905)
6.女金丸中牛皮源成分检查项补充检验方法(BJY201906)
7.女金丸中苋菜红、日落黄和亮蓝检查项补充检验方法(BJY201907)
8.女金丸中松香酸检查项补充检验方法(BJY201908)
9.洁白胶囊(丸)中松香酸检查项补充检验方法(BJY201909)
10.归脾丸(浓缩丸)中酸枣仁植物组织检查项补充检验方法(BJY201910)
11.胆香鼻炎片中苍耳子、金银花及甘草植物组织检查项补充检验方法(BJY201911)
12.阿胶颗粒中牛皮源成分检查项补充检验方法(BJY201912)
13.阿胶黄芪口服液中牛皮源成分检查项补充检验方法(BJY201913)
14.妇科止带片中牛皮源成分检查项补充检验方法(BJY201914)
15.鹿角胶中猪皮源成分检查项补充检验方法(BJY201915)
16.龟甲胶中猪皮源成分检查项补充检验方法(BJY201916)
17.阿胶中猪皮源成分检查项补充检验方法(BJY201917)
附件1
心可宁胶囊中酸性红73检查项补充检验方法(BJY 201901)
【检查】酸性红73 照高效液相色谱法(中国药典2015年版通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.05mol/L乙酸铵溶液(24∶76)为流动相;检测波长为509nm。理论板数按酸性红73峰计算应不低于3000。
对照品溶液的制备 取酸性红73对照品适量,精密称定,加70%甲醇制成每1ml中含20μg的溶液,即得。
供试品溶液的制备 取本品内容物4.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇20ml,密塞,称定重量,超声处理(功率500W,频率40kHz)20分钟,放冷,称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法 分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,即得。
结果判断 供试品色谱中,应不得出现与对照品色谱保留时间相同的色谱峰。若出现保留时间相同的色谱峰,采用二级管阵列检测器比较相应色谱峰在200~600nm波长范围内的紫外-可见吸收光谱,吸收光谱应不相同。
备注:必要时,可采用高效液相色谱-质谱联用方法验证。建议使用乙腈-0.01mol/L乙酸铵溶液流动相系统。
附件2
妇科止带片中金胺O检查项补充检验方法(BJY 201902)
【检查】金胺O照高效液相色谱法(中国药典 2015年版通则0512)测定。
色谱条件与系统适应性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.033mol/L磷酸二氢钾溶液(30∶70)为流动相;检测波长为435nm。理论板数按金胺O峰计算应不低于2000。
对照品溶液的制备 取金胺O对照品适量,加70%乙醇制成每1ml含2μg的溶液,即得。
供试品溶液的制备 取本品(包衣片除去包衣)适量,研细,取2.5g,加入70%乙醇10ml,密塞,超声处理30分钟,放冷,滤过,取续滤液,即得。
测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,记录色谱图。
结果判断 供试品色谱中,应不得出现与对照品色谱保留时间相同的色谱峰。若出现保留时间相同的色谱峰,则采用二极管阵列检测器比较相应色谱峰在320~500nm波长范围内的紫外-可见吸收光谱,吸收光谱应不相同。
备注:必要时,可采用高效液相色谱-质谱联用方法验证。建议采用乙腈-5mmol/L乙酸铵溶液(25∶75)流动相系统。
附件3
心宁片中赤芍、三七茎叶植物组织检查项补充检验方法(BJY 201903)
【检查】赤芍、三七茎叶植物组织 本品为部分浸膏片,除三七、川芎原药材组织外,不得检出赤芍、三七茎叶植物组织。
取本品2片,除去包衣,研细,取0.1g,加水10ml使溶解,离心,弃去上清液,沉淀加水2ml使混悬,加水合氯醛试液2ml,加热透化,摇匀,取混悬液1滴,置载玻片上,加稀甘油1滴,盖上盖玻片,置显微镜100倍下,参照下图位置,选取9个检查点检视。视野中不得检出以下任一植物组织:草酸钙簇晶众多,散在或存在于薄壁细胞中,常数个至数十个排列成行,直径7~41μm,棱角较平截或稍尖,有的似方晶状;木栓细胞淡红色、棕色或微显紫色,表面观呈长条形、长方形或长多角形,壁稍厚,平直或稍波状弯曲,有的细胞中充满棕色或红棕色块状物(赤芍)。茎纤维长,多成束存在;叶肉组织由类圆形薄壁细胞组成(三七茎叶)。
如仅有1个检查点视野中检出上述植物组织,应依法制片复试,复试应不得检出。
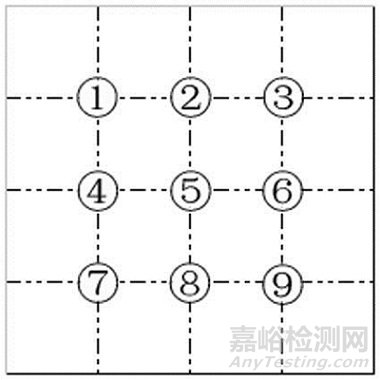
图:盖玻片上检查点示意图
附件4
银柴颗粒中灰毡毛忍冬皂苷乙检查项补充检验方法(BJY 201904)
【检查】灰毡毛忍冬皂苷乙 照高效液相色谱法(中国药典 2015年版通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A ,以水为流动相B,按下表中的规定进行梯度洗脱;蒸发光散射检测器检测。理论板数按灰毡毛忍冬皂苷乙峰计算应不低于5000。

对照品溶液的制备 取灰毡毛忍冬皂苷乙对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得。
供试品溶液的制备 取本品5袋内容物,研细,混匀,取2g,精密称定,置具塞锥形瓶中,精密加入加甲醇25ml,称定重量,超声处理30分钟(300W,50kHz),放冷,再称定重量,加甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法 分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。
结果判断 供试品色谱中,应不得出现与对照品色谱保留时间相同的色谱峰。
备注:必要时,可采用高效液相色谱-质谱联用方法验证。建议采用乙腈-0.1%甲酸流动相系统。
附件5
心可舒胶囊中人参皂苷Rb3检查项补充检验方法(BJY 201905)
【检查】人参皂苷Rb3 照高效液相色谱法(中国药典2015年版通则0512)和质谱法(中国药典2015年版通则0431)测定。
色谱、质谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(色谱柱内径2.1mm);以乙腈为流动相A,水为流动相B,按下表中的规定进行梯度洗脱,流速为0.3ml/min;柱温为40℃;采用质谱检测器,电喷雾负离子模式(ESI-),进行多反应监测(MRM),选择质荷比m/z 1077.7→783.4和m/z 1077.7→945.4作为检测离子对。

对照品溶液的制备取人参皂苷Rb3对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,精密量取适量,用20%乙腈稀释制成每1ml含4ng的溶液,即得。
供试品溶液的制备取本品10粒的内容物,研细,取约1.2g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,置80℃水浴中加热回流2小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液1ml,置100ml量瓶中,加20%乙腈稀释至刻度,摇匀,滤过,即得。
测定法分别精密吸取对照品溶液与供试品溶液各5µl,注入高效液相色谱-质谱联用仪,测定,记录色谱图。
判定原则(1)供试品的提取离子流色谱中,未同时出现与对照品溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时出现与对照品溶液色谱相应的色谱峰,且供试品色谱中m/z 1077.7→783.4的色谱峰面积值不大于对照品溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时出现与对照品溶液色谱相应的色谱峰,且供试品色谱中m/z 1077.7→783.4的色谱峰面积值大于对照品溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与对照品溶液色谱相应的色谱峰。
附件6
女金丸中牛皮源成分检查项补充检验方法(BJY 201906)
【检查】牛皮源成分 照高效液相色谱(中国药典2015年版通则0512)和质谱法(中国药典2015年版通则0431)测定。
色谱、质谱条件与系统适用性实验 以十八烷基硅烷键合硅胶为填充剂(色谱柱内径为2.1mm);以0.1%甲酸乙腈溶液为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱,流速为每分钟0.3m1。采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 641.3(双电荷)→726.2和m/z 641.3(双电荷)→783.3作为检测离子对。取牛皮源成分参比溶液,进样5µl,按上述离子对测定的MRM色谱峰的信噪比均应大于10:1。

牛皮源成分参比溶液的制备 取黄明胶对照药材0.10g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取上述溶液5ml,置100ml量瓶中,加入阿胶对照药材粉末0.20g,加1%碳酸氢铵溶液80ml,超声处理30分钟,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解12小时,即得。
供试品溶液的制备 取本品水蜜丸适量,研碎,取4.00g,精密称定;或取大蜜丸10丸,剪碎,混匀,取7.00g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟使溶散,加1%碳酸氢铵溶液稀释至刻度,摇匀,离心(3000转/分钟)5分钟,将上清液转移至另一离心管中,离心(9000转/分钟)5分钟,取上清液,用0.45μm微孔滤膜滤过,取续滤液,自“用0.22μm微孔滤膜滤过”起,照牛皮源成分参比溶液的制备方法同法操作,即得。
测定法 分别精密吸取参比溶液与供试品溶液各5μl,注入液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时出现与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时出现与参比溶液色谱离子质荷比(m/z)数值相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。
附件7
女金丸中苋菜红、日落黄和亮蓝检查项补充检验方法(BJY 201907)
【检查】苋菜红、日落黄和亮蓝(1)取本品2g,加硅藻土1g,研细,加50%甲醇20ml,超声处理30分钟,滤过,滤液作为供试品溶液。另取苋菜红、日落黄和亮蓝对照试剂,加50%甲醇制成每1ml含20μg的混合溶液,作为对照试剂溶液。照薄层色谱法(中国药典2015年版通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-正丁醇-乙醇-浓氨试液-水(1∶3∶3∶1∶1)为展开剂,展开,取出,晾干,日光下检视。供试品色谱中,在与对照试剂色谱相应的位置上,不得检出相同颜色的斑点;若检出相同颜色的斑点,或相同位置有干扰不能判断时,则采用下列高效液相色谱法验证。
(2)照高效液相色谱法(中国药典2015年版通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相A,以0.05mol/L乙酸铵为流动相B,按下表梯度进行洗脱;采用二极管阵列检测器;检测波长为521nm(检测苋菜红)、484nm(检测日落黄)、629nm(检测亮蓝)。理论板数按苋菜红峰计算应不低于4000。
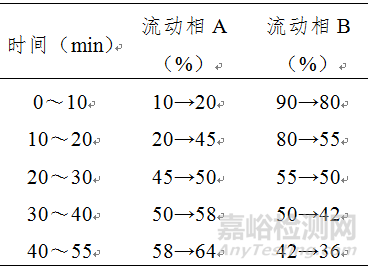
测定法 分别精密吸取供试品溶液和对照试剂溶液各10μl,注入液相色谱仪,测定,即得。
结果判断 供试品色谱中,应不得出现与对照试剂色谱保留时间相同的色谱峰。若出现保留时间相同的色谱峰,则采用二极管阵列检测器比较相应色谱峰在200~700nm波长范围内的紫外-可见吸收光谱,吸收光谱应不相同。
备注:必要时,可采用高效液相色谱-质谱联用方法验证。
附件8
女金丸中松香酸检查项补充检验方法(BJY 201908)
【检查】松香酸 照高效液相色谱法(中国药典2015年版通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%甲酸(80 :20)为流动相;采用二极管阵列检测器;检测波长为241nm。理论板数按松香酸峰计算应不低于2000。
对照溶液的制备(临用新制) 取松香酸对照试剂适量,精密称定,加50%甲醇制成每1ml含2μg的溶液,作为对照试剂溶液。另取11-羰基-β-乙酰乳香酸对照品适量,精密称定,加50%甲醇制成每1ml含2μg的溶液,作为参照溶液。
供试品溶液的制备 取本品9g,加硅藻土4.5g,研细,取约3g,精密称定,加50%甲醇20ml,超声处理30分钟,滤过,精密量取续滤液1ml,置10ml量瓶中,加50%甲醇稀释至刻度,摇匀,即得。
测定法 分别精密吸取供试品溶液、对照试剂溶液与参照溶液各10μl,注入液相色谱仪,记录色谱图。
结果判断 供试品色谱中,在与松香酸对照试剂溶液色谱峰保留时间相应的位置上不得出现相同的色谱峰。若出现保留时间相同的色谱峰,采用二极管阵列检测器比较相应色谱峰的紫外-可见吸收光谱,吸收光谱应不同(松香酸对照试剂色谱峰在241nm显示最大吸收);若吸收光谱相同,且该色谱峰的峰面积大于11-羰基-β-乙酰乳香酸参照溶液色谱峰的峰面积值,则视为阳性检出。
备注:必要时,可采用高效液相色谱-质谱联用方法验证。
附件9
洁白胶囊(丸)中松香酸检查项补充检验方法(BJY 201909)
【检查】松香酸 (1)取本品胶囊内容物适量,研细,取1g;取本品丸适量,研细,取1g,加乙醇20ml,密塞,超声处理20分钟,滤过,蒸干,残渣加无水乙醇2ml溶解,作为供试品溶液。取松香酸对照试剂适量,加乙醇制成每1ml含1mg的溶液,即得(临用新制)。照薄层色谱法(中国药典2015年版通则0502)试验,吸取供试品溶液和对照试剂溶液各5μl,分别点于同一硅胶GF254薄层板上,以甲苯-乙酸乙酯-正庚烷-无水甲酸(8:2:1:0.3)为展开剂,展开,取出,晾干,放置10分钟后置紫外光灯(254nm)下检视。供试品色谱中,在与对照试剂色谱相应的位置上,不得显相同的荧光淬灭斑点。再喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与松香酸对照试剂相应的位置上,不得显相应的荧光斑点。若出现相同颜色的斑点,则用下列高效液相色谱法验证。
(2)照高效液相色谱法(中国药典2015年版通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%甲酸溶液(80:20)为流动相;检测波长为241nm。理论板数按松香酸峰计算应不低于2000。
对照试剂溶液的制备(临用新制) 取松香酸对照试剂适量,加乙醇制成每1ml含10μg的溶液,即得。
供试品溶液的制备 取本品胶囊内容物适量,研细,取0.2g;取本品丸适量,研细,取0.2g,加乙醇20ml,超声处理20分钟,放至室温,上清液用微孔滤膜(0.45μm)滤过,取续滤液,即得。
测定法 分别吸取供试品溶液和对照试剂溶液各10μl,注入液相色谱仪,记录色谱图。
结果判断供试品色谱中,在与松香酸对照试剂溶液色谱峰保留时间相应的位置上不得出现相同的色谱峰。若出现保留时间相同的色谱峰,则采用二极管阵列检测器比较相应色谱峰的紫外-可见吸收光谱,吸收光谱应不相同(松香酸对照试剂色谱峰在241nm显示最大吸收);若吸收光谱相同,则视为阳性检出。
备注:必要时,可采用高效液相色谱-质谱联用方法进行验证。
附件10
归脾丸(浓缩丸)中酸枣仁植物组织检查项补充检验方法(BJY 201910)
【检查】酸枣仁植物组织 本品为部分浸膏浓缩丸,除党参、当归、甘草、木香植物组织特征外,不得检出酸枣仁植物组织。
取本品5丸,研细,称取0.1g,加水10ml超声使溶解,离心(转速为每分钟2000转)10分钟,弃去上清液,沉淀加水2ml使混悬,吸取混悬液1滴于载玻片上,加水合氯醛试液适量,加热透化,盖上盖玻片,置显微镜100倍下,参照下图位置,选取9个检查点检视,视野中不得检出以下任一植物组织: 种皮栅状细胞棕红色,表面观多角形,直径约15μm,壁厚,木化,胞腔小,侧面观呈长条形,外壁增厚,侧壁上中部甚厚,下部渐薄,底面观类多角形或圆多角形;种皮内表皮细胞棕黄色,表面观长方形或类方形,垂周壁连珠状增厚,木化。
如仅有1个检查点视野中检出上述植物组织,应依法制片复试,复试不得检出。
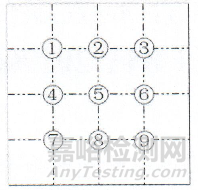
图:盖玻片上检查点示意图
附件11
胆香鼻炎片中苍耳子、金银花及甘草植物组织检查项补充检验方法(BJY 201911)
【检查】苍耳子、金银花及甘草植物组织 本品为部分浸膏片,除鹅不食草植物组织特征外,不得检出苍耳子、金银花及甘草植物组织。
取本品2片,研细,称取0.1g,加水10ml超声使溶解,离心(转速为每分钟2000转)10分钟,弃去上清液,沉淀加水2ml使混悬,取混悬液1滴置载玻片上,加水合氯醛试液适量,加热透化,盖上盖玻片,置显微镜100倍下,参照下图位置,选取9个检查点检视,视野中不得检出以下任一植物组织:总苞纤维成束,常呈纵横交叉排列。果皮表皮细胞棕色,类长方形,常与下层纤维相连(苍耳子)。花粉粒类圆形或三角形,表面具细密短刺及细颗粒状雕纹,具3孔沟。草酸钙簇晶直径6~45μm,存在于薄壁细胞中(金银花)。纤维成束,直径8~14μm,壁厚,微木化,周围薄壁细胞含草酸钙方晶,形成晶纤维。具缘纹孔导管较大(甘草)。
如仅有1个检查点视野中检出上述植物组织,应依法制片复试,复试不得检出。
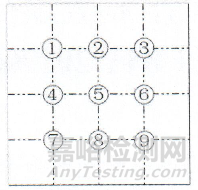
图:盖玻片上检查区域示意图
附件12
阿胶颗粒中牛皮源成分检查项补充检验方法(BJY 201912)
【检查】牛皮源成分照高效液相色谱法(中国药典2015年版通则0512)和质谱法(中国药典2015年版通则0431)测定。
色谱、质谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(色谱柱内径2.1mm);以乙腈为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为每分钟0.3ml;采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 641.3(双电荷)→726.2和m/z 641.3(双电荷)→783.3作为检测离子对。取牛皮源成分参比溶液,进样5μl,按上述离子对测定的MRM色谱峰的信噪比均应大于10:1。

牛皮源成分参比溶液的制备取黄明胶对照药材0.12g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取上述溶液5ml,置100ml量瓶中,并加入阿胶对照药材粉末0.24g,加1%碳酸氢铵溶液80ml,超声处理30分钟,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解12小时,即得。
供试品溶液的制备取阿胶颗粒粉末0.20g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,加1%碳酸氢铵溶液稀释至刻度,摇匀,自“用0.22μm微孔滤膜滤过”起,照牛皮源成分参比溶液的制备方法同法操作,即得。
测定法分别吸取牛皮源成分参比溶液与供试品溶液各5μl,注入高效液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时出现与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。
附件13
阿胶黄芪口服液中牛皮源成分检查项补充检验方法(BJY 201913)
【检查】牛皮源成分高效液相色谱法(中国药典2015年版通则0512)和质谱法(中国药典2015年版通则0431)测定。
色谱、质谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(色谱柱内径2.1mm);以乙腈为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为每分钟0.3ml;采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 641.3(双电荷)→726.2和m/z 641.3(双电荷)→783.3作为检测离子对。取牛皮源成分参比溶液,进样5μl,按上述离子对测定的MRM色谱峰的信噪比均应大于10:1。

牛皮源成分参比溶液的制备取黄明胶对照药材0.10g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取上述溶液5ml,置100ml量瓶中,并加入阿胶对照药材粉末0.20g,加1%碳酸氢铵溶液80ml,超声处理30分钟,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解12小时,即得。
供试品溶液的制备精密量取阿胶黄芪口服液1ml,置50ml量瓶中,加1%碳酸氢铵溶液稀释至刻度,摇匀,自“用0.22μm微孔滤膜滤过”起,照牛皮源成分参比溶液的制备方法同法操作,即得。
测定法分别吸取牛皮源成分参比溶液与供试品溶液各5μl,注入高效液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时出现与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。
附件14
妇科止带片中牛皮源成分检查项补充检验方法
(BJY 201914)
【检查】牛皮源成分 照高效液相色谱法(中国药典2015年版通则0512)和质谱法(中国药典2015年版通则0431)测定。
色谱、质谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(色谱柱内径为2.1mm);以乙腈为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱,流速为每分钟0.3ml;采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 641.3(双电荷)→726.2和m/z 641.3(双电荷)→783.3作为检测离子对。取牛皮源成分参比溶液,进样5μl,按上述离子对测定的MRM色谱峰的信噪比均应大于10∶1。

牛皮源成分参比溶液的制备 取黄明胶对照药材0.10g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理60分钟,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取上述溶液5ml,置100ml量瓶中,加入阿胶对照药材粉末0.20g,加1%碳酸氢铵溶液80ml,超声处理30分钟,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100 μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解20小时,即得。
供试品溶液的制备 取妇科止带片10片(包衣片除去包衣),精密称定,研细,混匀,取约1片的重量(约相当于含胶类药材0.2g),精密称定,置100ml量瓶中,加1%碳酸氢铵溶液80ml,超声处理60分钟,加1%碳酸氢铵溶液稀释至刻度,摇匀,自“用0.22μm微孔滤膜滤过”起,照牛皮源成分参比溶液的制备方法同法操作,即得。
测定法 分别吸取牛皮源成分参比溶液与供试品溶液各5μl,注入高效液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时出现与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时出现与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 641.3(双电荷)→726.2的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。
备注:建议采用固定相粒径不大于3μm规格的色谱柱。
附件15
鹿角胶中猪皮源成分检查项补充检验方法(BJY 201915)
【检查】猪皮源成分照高效液相色谱法(中国药典2015年版四部通则0512)和质谱法(中国药典2015年版四部通则0431)测定。
色谱、质谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(色谱柱内径2.1mm);以乙腈为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱,流速为每分钟0.3ml;采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 774.5(双电荷)→977.8和m/z 774.5(双电荷)→1034.6作为检测离子对。取猪皮源成分参比溶液,进样5μl,按上述离子对测定的MRM色谱峰的信噪比均应大于10:1。

猪皮源成分参比溶液的制备取新阿胶对照药材0.10g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,使样品完全溶解,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取上述溶液5ml置100ml量瓶中,并加入鹿角胶对照药材粉末0.20g,加1%碳酸氢铵溶液80ml,超声处理30分钟,使样品完全溶解,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解12小时,即得。
供试品溶液的制备取本品粉末0.10g,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,使样品完全溶解,加1%碳酸氢铵溶液稀释至刻度,摇匀,自“用0.22μm微孔滤膜滤过”起,照猪皮源成分参比溶液的制备方法同法操作,即得。
测定法分别吸取猪皮源成分参比溶液与供试品溶液各5μl,注入高效液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时检出与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时检出与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 774.5(双电荷)→977.8的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时检出与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 774.5(双电荷)→977.8的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。
附件16
龟甲胶中猪皮源成分检查项补充检验方法(BJY 201916)
【检查】猪皮源成分照高效液相色谱法(中国药典2015年版四部通则0512)和质谱法(中国药典2015年版四部通则0431)测定。
色谱、质谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(色谱柱内径2.1mm);以乙腈为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱,流速为每分钟0.3ml;采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 774.5(双电荷)→977.8和m/z 774.5(双电荷)→1034.6作为检测离子对。取猪皮源成分参比溶液,进样5μl,按上述离子对测定的MRM色谱峰的信噪比均应大于10:1。

猪皮源成分参比溶液的制备取新阿胶对照药材0.10g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,使样品完全溶解,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取取上述溶液5ml,置100ml量瓶中,加入龟甲胶对照药材粉末0.20g,加1%碳酸氢铵溶液80ml,超声处理30分钟,使样品完全溶解,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解12小时,即得。
供试品溶液的制备取本品粉末0.10g,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,使样品完全溶解,加1%碳酸氢铵溶液稀释至刻度,摇匀,自“用0.22μm微孔滤膜滤过”起,照猪皮源成分参比溶液的制备方法同法操作,即得。
测定法分别吸取猪皮源成分参比溶液与供试品溶液各5μl,注入高效液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时检出与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时检出与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 774.5(双电荷)→977.8的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时检出与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 774.5(双电荷)→977.8的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。
附件17
阿胶中猪皮源成分检查项补充检验方法(BJY 201917)
【检查】猪皮源成分照高效液相色谱法(中国药典2015年版四部通则0512)和质谱法(中国药典2015年版四部通则0431)测定。
色谱、质谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(色谱柱内径2.1mm);以乙腈为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱,流速为每分钟0.3ml;采用质谱检测器,电喷雾正离子模式(ESI+),多反应监测(MRM),选择m/z 774.5(双电荷)→977.8和m/z 774.5(双电荷)→1034.6作为检测离子对。取猪皮源成分参比溶液,进样5μl,按上述离子对测定的MRM色谱峰的信噪比均应大于10:1。

猪皮源成分参比溶液的制备取新阿胶对照药材0.10g,精密称定,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,使样品完全溶解,加1%碳酸氢铵溶液稀释至刻度,摇匀。精密量取上述溶液5ml置100ml量瓶中,加入阿胶对照药材粉末0.20g,加1%碳酸氢铵溶液80ml,超声处理30分钟,使样品完全溶解,再加1%碳酸氢铵溶液稀释至刻度,摇匀,用0.22μm微孔滤膜滤过,取续滤液100μl,置微量进样瓶中,加胰蛋白酶溶液(取序列分析级胰蛋白酶,加1%碳酸氢铵溶液制成每1μl中含1μg的溶液,临用前现配)10μl,摇匀,37℃恒温酶解12小时,即得。
供试品溶液的制备取本品粉末0.10g,置50ml量瓶中,加1%碳酸氢铵溶液40ml,超声处理30分钟,使样品完全溶解,加1%碳酸氢铵溶液稀释至刻度,摇匀,自“用0.22μm微孔滤膜滤过”起,照猪皮源成分参比溶液的制备方法同法操作,即得。
测定法分别吸取猪皮源成分参比溶液与供试品溶液各5μl,注入高效液相色谱-质谱联用仪,测定,即得。
判定原则(1)供试品的提取离子流色谱中,未同时检出与参比溶液色谱相应的色谱峰,视为未检出;(2)供试品的提取离子流色谱中,同时检出与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 774.5(双电荷)→977.8的色谱峰面积值不大于参比溶液中相应的峰面积值者,视为未检出;(3)供试品的提取离子流色谱中,同时检出与参比溶液色谱相应的色谱峰,且供试品色谱中m/z 774.5(双电荷)→977.8的色谱峰面积值大于参比溶液中相应的峰面积值者,视为检出。
结果判定供试品的提取离子流色谱中,应不得检出与参比溶液色谱相应的色谱峰。

来源:国家药监局


