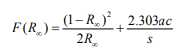考虑到药物晶型是影响药品质量的重要因素之一,所以仅定性分析原料药或制剂中的晶型难以满足药品的质量控制要求。为了测定原料药或制剂中有效晶型的含量, 从而确保制剂的疗效, 建立适当的多晶型定量方法不可或缺,。另一方面,当药物的多晶型或溶剂化物之间的理化性质相差很大, 以致于影响到药物制剂的质量(如生物利用度或化学稳定性)或制剂生产的重现性时, 建立多晶型的定量方法必不可少。比如一种降血糖药物那格列奈存在三种主要晶型——H晶型、S晶型和B晶型.其中H晶型和S晶型被认定为降血糖的有效晶型,而B晶型的热稳定性和溶解性较差,应严格控制,所以研究者们往往对那格列奈中以上三种晶型均进行定量分析,以便保证药物的安全和疗效。再者,在知识产权方面, 具有与原研药的晶型生物等效甚至药效更高的新晶型, 可以作为一个附加专利, 以便延长药物的专利保护期。综上所述,建立药物晶型的定量方法为药物研发和生产过程中的重要环节。本文笔者总结了几种常见的药物晶型定量方法,旨在为广大研究者选择晶型定量方法时提供思路。
一、粉末X射线衍射法(XRPD)
XRPD是最常用的药物多晶型定量方法,通过测量药物晶体对X射线的衍射模式来确定晶型的种类和含量。该方法专属性最强,通过晶型定量可直观地获得晶型变化、结晶度等信息。XRPD常用的定量方式分为两种——单峰法和全谱法。
1.1 单峰法
单峰法的原理是同一药物混晶分析的情况下,特定衍射峰的强度与某一晶型的浓度呈正比,衍射强度对浓度(质量分数)回归可以得到线性回归曲线。然而,运用此曲线定量分析制剂中的晶型含量时, 需考虑到原料药与辅料之间质量吸收系数的差异可能会使回归曲线偏离线性。根据《中国药典》四部通则0451,单峰法又可细分为外标法、内标法和标准加人法,此方法在药物晶型定量方面应用最广泛。周新波等用单峰法对埃索美拉唑镁盐的多晶型进行定量分析,他们将三水合物晶型和二水合物A晶型的峰面积比值为定量参数,建立标准曲线,结果表明:两种晶型的特征衍射峰的峰面积比值与三水合物的含量呈良好的线性关系,在三水合物含量为 3 %~15 % 的范围内,相关系数 r = 0. 9996,该法实现了对埃索美拉唑镁盐中三水合物晶型和二水合物 A 晶型的准确定量分析。徐婷等同样用单峰法对盐酸文拉法辛的多晶型进行定量分析,他们也是将晶型2的特征衍射峰和晶型6的特征衍射峰的峰面积比值为定量参数,建立标准曲线,测定盐酸文拉法辛原料中晶型2和晶型6 的含量。结果:两特征衍射峰峰面积比值,与晶型6的含量呈良好的线性关系,在晶型6含量为 5 %~95 %的范围内,得到的标准曲线为相关系数 r = 0 999;方法重复性RSD为1.86 %;定量限为5.02 %,该方法准确可靠、重复性良好,可用于盐酸文拉法辛原料中晶型2和晶型6的定量分析。
单峰法对样品的信息要求较小、方法简单。但是,该定量方法灵敏度很难满足微量或痕量晶型的定量分析,且定量时严重依赖标准品的含量,而且制样时易产生的择优取向效应会严重影响单峰法的定量准确度。因此,在实际应用中应严格控制操作条件才能得到较可靠的数据。有研究者为减弱或消除择优取向的不良影响,进行校正,比如Tiwari等通过建立标准曲线和对仪器、操作参数的二维最优化处理,成功实现了对奥氮平多晶型的定量分析,方法检测限和定量限分别为0.40 %和1.22 %,灵敏度极高。
1.2 全谱图法
全谱图法是通过建立XRPD全谱图与晶型含量之间的关系进行分析的方法。与单峰法相比,全谱图法需要更多的关于待分析物的信息。目前,全谱图法主要有偏最小二乘法(PLS)、全粉末谱图分解法(WPPD)和Rietveld法。
以化学计量学为基础的全谱法可以通过分析布拉格衍射和弥漫散射来定量样品, 因而其信噪比、灵敏度和专属性明显提高。Moore等分别以经典最小二乘回归法 (CLS)、主成分回归法(PCR)和偏最小二乘法(PLS)为基础, 利用全谱法定量分析了两种晶态样品和两种无定型样品。模型比较表明,PLS回归为较理想的定量模型。Croker等也利用全谱图PLS模型建立了吡拉西坦晶型Ⅱ和晶型Ⅲ的定量分析方法。
WPPD 法的定量原理是将积分强度参数、晶胞参数及峰型参数等由最小二乘法结合某一晶型的可信度因子进行修正,以提高晶型定量准确度。Uvarov等应用WPPD法对双硫酸氯吡格雷二元混晶成功定量。他们将衍射图分解为峰型方程和本底方程,通过CLS 使某一纯晶型的衍射数据与上述方程计算所得的数据相拟合。每一组分的可信度因子通过 CLS 拟合,直到与未知组分的图谱达到最好的吻合。马乐伟等利用此法和单峰法分别对硫酸氢氯吡格雷二元混晶进行定量, 所得数据结果表明, WPPD法具有更高的准确度, 方法定量限在1.0 %~1.5 %。
Rietveld 法的原理本质上与WPPD 法相同,但是需要结构模型计算特定相的峰强度,即事先假设一种晶体结构模型和结构参数, 在此基础上,结合某种峰型参数计算晶体的衍射图谱,通过计算机程序,逐点比较衍射强度的计算值与实验值, 利用PLS法调节晶体结构和峰型参数,使计算图谱与实验图谱之间的加权剩余方差因子最小。Nemet等通过Rietveld法使用Fullprof软件建立了法莫替丁晶型A和晶型B混合物的定量分析方法。该方法首先结合纯组分的XRPD谱图得到计算模型,再通过质量分数为50%的晶型A和50%晶型B混合物对模型进行修正后即可用于晶型的定量分析。结果显示,晶型A和晶型B两者的含量计算值与实际值的标准曲线相关系数(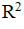 )分别达到0.9999和0.9940,体现了较好的吻合性,说明该定量分析方法可靠性较高。与WPPD 法相比,Rietveld法尽管准确度更高,但是需要更多的晶格参数信息,除了需要质量吸收系数、结构因子外, 还需要原子坐标、位置占有率等用于分析结构因子的参数。因此,当所得的晶体结构信息很少时,Rietveld法不如WPPD法有优势。
)分别达到0.9999和0.9940,体现了较好的吻合性,说明该定量分析方法可靠性较高。与WPPD 法相比,Rietveld法尽管准确度更高,但是需要更多的晶格参数信息,除了需要质量吸收系数、结构因子外, 还需要原子坐标、位置占有率等用于分析结构因子的参数。因此,当所得的晶体结构信息很少时,Rietveld法不如WPPD法有优势。
与单峰法相比,全谱图法不需要标准品,降低了试样粒度和择优取向等对定量分析的影响,但该方法需要较多的晶格参数信息,且方法建立及计算过程较为复杂,需借助特殊软件才可实现定量分析。
二、差示扫描量热法(DSC)
DSC是一种测量药物晶体在加热或冷却过程中吸收或释放热量变化的方法,晶型转变或熔融时会发生热效应,相应的热量变化与晶型含量之间存在比例关系,故可以用于测定晶型的含量。另外,在保证固态药物100 %晶态的前提下,DSC还可以用于测定药物的结晶度。Sheikhzadeh等采用DSC法开发了丁螺环酮盐酸盐晶型Ⅰ和晶型Ⅱ二元混合物的定量分析方法,分别以晶型Ⅰ含量对两种晶型特征峰峰高的比值与晶型Ⅰ的特征峰峰面积做图,发现两者均存在良好的线性关系,但峰面积的准确度更高。杨敏等采用DSC法定量分析固体分散体中的依折麦布晶型,通过在辅料中加入不同比例的晶型药物,制备不同含量晶型药物的固体分散体样品,建立其吸热峰焓值与晶型药物含量间的线性关系。结果表明,当固体分散体中晶型药物含量为0.5 %~10 %时,其晶型含量与吸热峰焓值间的线性关系良好,且方法具有良好的精密度,可作为检测 1 固体分散体中无定形药物转化为晶型药物的定量分析方法。
除了传统的DSC法,还包括加强版的温度调幅式DSC(MTDSC)和高效DSC(HSDSC)。MTDSC 是在线性加热外叠加一个正弦振荡方式的加热。缓慢的线性加热时,可得到较高的分辨率;而正弦波振荡方式加热时, 造成了瞬间的温度剧烈变化,故灵敏度较高。MTDSC克服了传统的DSC不能同时具备高灵敏度和高分辨率的不足,配合傅里叶变换方法将总热流分解成可逆成分和不可逆成分,从而将许多重叠的转变峰分开。由于该法对无定型的玻璃态转变温度较为敏感, 因此多用于无定型药物或辅料的定量分析。Guinot等利用纯无定型 在玻璃化转变温度时热容量的跳动值建立了标准曲线, 成功地定量了X 化合物微粉化样品的结晶度, 其检测限和定量限分别低至0.9 %和3.0 %。Tong等也利用MTDSC 定量分析了昔萘酸沙美特罗中晶型Ⅱ杂质的含量,定量限低于1 %。
传统的DSC在测定很低含量的无定型药物方面具有很大的困难,而HSDSC因高热流率而提高了其检测的灵敏度和准确度,而且 HSDSC的加热和冷却速率可达到500 ℃/min。 亚稳态或无定型属于热力学不稳定体系, 在较快的升温速率下可以降低测量过程中晶型的转变。因此, 该法的突出优势在于分析样品中少量的亚稳态或无定型组分,尤其是无定型含量低于10 %的样品。Gabbott等用无定型乳糖验证HSDSC的性能,得出 HSDSC可以测试含量低于1.5 %的样品,具有很高的灵敏度。McGregor等采用HSDSC法以250 ℃/min升温速率定量测得卡马西平混晶样品中晶型的相对含量, 得出卡马西平晶型Ⅱ熔融焓的检测限能够达到1 %,但是由于晶种引起的部分重结晶,严重限制了其定量方面的应用。
DSC法所需样品量少,操作简单,但是测试过程对样品的损害是不可逆的,因而不适于定量分析不易获得或贵重样品。晶型的热力学差异在测量时受样品粒径、厚度、样品盘位置及热电偶等因素的影响较大,加之不同样品本身的热力学差异,往往导致不同样品用同一定量方法的检测限和定量限存在较大差异。
三、红外光谱法(IR)
IR是一种通过测量药物晶体对红外光的吸收谱来确定晶型的方法,不同晶型的药物会有不同的吸收峰。通过峰形、峰位和峰强度判断是否存在晶型差异,进行晶型鉴别和定量。目前用于晶型定量分析的红外光谱法主要有衰减全反射-傅里叶变换红外光谱法(ATR-FTIR)、漫反射傅里叶变换红外光谱法(DRIFTS)和近红外光谱法(NIRS)。
3.1 ATR-FTIR
ATR-FTIR的原理是当入射角大于临界角时,光会在样品与ATR晶体交界面上发生全反射。在全反射的过程中,光会在界面上形成一个电磁场,这个电磁场称为表面等离子体波(Surface Plasmon Polariton, SPP)。当样品与ATR晶体接触时,样品中的化学物质会吸收特定波长的红外光,产生特征性的振动光谱。1998年,Salari等首先将对于3种晶型,实际值和计算值之间均具有良好的线性关系,校正系数分别为0.981,0.982,0.986。这说明ATR-FTIR法与PLS模型相结合可成功地应用于药物多晶型的定量分析。ATR-FTIR法具有检测快速、对试样无损害、试样处理简单、可避免相转变发生等优点,因此在药物晶型定量分析方面具有很好的应用前景。
3.2 DRIFTS
DRIFTS法提高了红外光谱法定量分析药物晶型的准确度。一般情况下,DRIFTS的相对强度与样品浓度不成比例, 不能以Lambert-Beer 定律定量,但可通过Kubelka-Munk 方程计算:
式中,F(R∞): K-M 函数;R∞: 样品层无限厚时的反射率;a: 样品的摩尔吸收系数;c: 样品浓度;s: 散射系数。
由上述方程可知, 为了获得 F(R∞) 与c的线性关系, 需要保证散射系数s为一常量。在被测物浓度较低时, 散射系数主要取决于无吸收的分散剂(如 KBr),此时可以用 Lambert-Beer 定律定量。但浓度较大时,散射系数发生变化而使 Lambert-Beer定律不再适用,此时需用Kubelka-Munk 方程。
为了减小散射系数对实验结果的影响, 可以通过控制样品粒子的均一性、堆密度、粒径大小和形状来实现。此外,实际应用中往往引入化学计量法。Braga 等利用DRIFT结合 PLS,定量分析了卡马西平中混晶的相对含量。方法验证结果表明,此法可以作为 PXRD 的替代性方法;Kachrimanis 等利用DRIFTS与ANN相结合定量分析了甲苯达唑 A-C 混晶样品, 通过 ANN 模型对二次导数IR谱图的拟合处理,得到了较好的实验结果。与 PLS 模型相比,ANN 模型具有明显的优势。
利用 DRIFTS 法进行晶型定量时,由于样品分散在无吸收的介质内,样品制备过程不受热能和机械能的影响。因此,避免了制备过程中晶型的转变;此外,DRIFTS对粒径大小的影响不敏感、仪器通用性较强,使得DRIFTS非常适于晶型的定量,尤其适于晶型之间转 化规律及转化量的研究。
3.3 NIRS
NIRS是指波长介于可见光与中红外区之间的电磁波,美国材料试验学会规定其波长范围为 780 nm~2526 nm。NIRS定量分析主要是应用化学计量学方法通过提取试样光谱与试样含量之间的NIRS相关信息,建立相关的数学校正模型,通过所得数学模型预测未知组分的含量或性质,原理上与XRPD的全谱图法类似。与DRIFTS相比,NIRS谱峰包含的信息少、分辨率低、信息谱带比较宽,有时还会有峰的重叠现象。为了增加相邻峰的分辨率,在数据计算前,往往对NIRS 光谱进行一阶导数或二阶导数处理;另外,一些光谱预处理技术,如标准正态变量变换 (standard normal variate, SNV)、多元散射校正(multivariate scatter correction, MSC)等可以更有效地减小由样品粒子大小差异引起的散射效应;此外,引入多元 线性回归 (multiple linear regression, MLR)、主成分分析法 (principal component analysis, PCA) 和 PLS等化学计量学模型也是解决此类问题的方法。Patel等利用NIRS定量分析了磺胺噻唑二元混晶系统中晶型Ⅰ和晶型Ⅱ, 分别以MLR、PLS 等为回归模型建立了磺胺噻唑晶型I的质量分数与二次导数光谱参数的回归曲线,其检测限达到0.3 %;Blanco等采用NIRS快速有效地鉴别阿奇霉素晶型并对其定量分析,同时还测定了样品中无定型的含量。此外,他们还结合PLS成功地定量分析了药物制剂中米卡霉素晶型的含量。
NIRS的显著特点是分析速度快,其快速性不仅可以鉴别活性成分的不同晶型,还可以检测原料药和制剂中晶型的纯度。因此在药物晶型定量方面具有良好的应用前景。
四、拉曼光谱法(Raman)
Raman光谱法是将单色光源照射到试样上,光子与试样分子振动的相互作用使得散射光的波长偏离入射光波长,对散射光进行收集分析的一种技术。因此,通过检测分子振动的Raman光谱特征频率可以分析试样的化学结构。对于同一种化合物的不同晶型,由于其分子堆积方式的不同导致分子振动不同,因此,不同晶型具有不同特征的Raman光谱。与IR类似,Raman也是通过峰形、峰位和峰强度判断是否存在晶型差异,进行晶型鉴别和定量。Raman法可与IR法形成互补。Roberts等采用Raman光谱法研究了甘露糖醇δ晶型和β晶型二元混合物的定量分析方法。起初选取的两种晶型的特征峰并未完全分开,原因是某一晶型会对另一晶型的特征峰强度造成干扰。为解决此问题,他们用混合物试样的特征峰强度之比进行校正,得到的β晶型含量计算值与实际值之间呈现良好的线性关系,相关系数达到0.9986,定量限低至0.5 %。此外,Kachrimanis等采用Raman光谱法与PLS模型相结合建立了扑热息痛晶型Ⅰ和晶型Ⅱ混合物的定量分析方法,并比较了3种预处理算法(正交信号校正(OSC)、标准正态变换和多元散射校正)对定量分析准确性的影响,比较结果表明,OSC的修正效果最好。
Raman光谱法对于试样的处理较少,一定程度上避免了转晶的发生,且允许在一定湿度 条件下进行测试,测试速度较快,因此具有较好的应用前景。但在某些情况下,不同晶型的Raman光谱可能会比较相近,故不容易找到独立且明显的特征峰。
五、固态核磁共振波谱法(ss-NMR)
ss-NMR是一种通过测量药物晶体中原子核的共振频率来确定晶型的方法。不同晶型,分子排列的差异,导致不同晶型间分子内相同原子的化学环境产生差异,表现为化学位移不同。通过测定不同化学位移处的峰强度, 可以定量分析药物多晶型。常规的NMR定量通常采用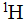 谱,但晶型定量与之不同,由于谱各向同性化学位移的范围只有12 ppm,质子−质子偶合作用引起的峰宽效应会导致谱的重叠, 严重限制了谱在定量方面的应用。相比之下,
谱,但晶型定量与之不同,由于谱各向同性化学位移的范围只有12 ppm,质子−质子偶合作用引起的峰宽效应会导致谱的重叠, 严重限制了谱在定量方面的应用。相比之下,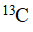 谱各向同性化学位移的范围明显扩大,而且高分辨率的谱较易获得。因此,谱更适于晶型定量。此外, 能给出高分辨率光谱的
谱各向同性化学位移的范围明显扩大,而且高分辨率的谱较易获得。因此,谱更适于晶型定量。此外, 能给出高分辨率光谱的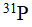 和
和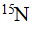 同样适于药物晶型的定量。Vickery等利用ss-NMR法建立了罗西非班晶型Ⅰ和晶型Ⅱ混合物的定量分析方法。他们选择了两组特征峰,并分别用特征峰之比进行校正。结果证明,两种方法的理论值与实际值均十分接近,说明该定量分析方法有效,可用于药物多晶型含量的质控。Tozuka等利用ss-NMR法建立了克拉霉素晶型Ⅰ和晶型Ⅱ混合物的定量分析方法。由于两种晶型的特征峰距离较近,略有交叉,因此使用软件对其进行分峰处理,并用晶型含量对特征峰面积之比做图,发现两者存在良好的线性关系,线性相关系数为0.99,可用于定量。
同样适于药物晶型的定量。Vickery等利用ss-NMR法建立了罗西非班晶型Ⅰ和晶型Ⅱ混合物的定量分析方法。他们选择了两组特征峰,并分别用特征峰之比进行校正。结果证明,两种方法的理论值与实际值均十分接近,说明该定量分析方法有效,可用于药物多晶型含量的质控。Tozuka等利用ss-NMR法建立了克拉霉素晶型Ⅰ和晶型Ⅱ混合物的定量分析方法。由于两种晶型的特征峰距离较近,略有交叉,因此使用软件对其进行分峰处理,并用晶型含量对特征峰面积之比做图,发现两者存在良好的线性关系,线性相关系数为0.99,可用于定量。
与其他光谱法(IR、Raman)相比,ss-NMR具有较高分辨率。但由于高速旋转通常用于收集高分辨率的ss-NMR 光谱,此技术不适用于可能经历相转变的亚稳态晶型的研究。而且,弛豫时间对固态药物中分子的移动性比较敏感,而温度的变化也会导致数值剧烈的变动,因而,实验过程中需要严格控制样品的温度。另外,信号累加所需的循环弛豫时间使此方法 测量时间较长,严重制约了此技术的常规应用。
六、热重分析法(TGA)和单晶X射线衍射法(XRSD)
TGA是一种测量药物晶体在加热过程中失去的质量变化的方法,可用于确定晶型物质中是否含有结晶溶剂或结晶水并进行定量。XRSD通常也用于测定晶型物质中结晶溶剂或结晶水的含量。以上两种方法在药物晶型定量方面具有较大的局限性,往往都是通过测得的结晶溶剂或结晶水的含量,进而推算晶型的含量,且仅适用于单一晶型,故在药物晶型定量实际应用中更多的作为辅助手段。此外,TGA法要求晶型的热稳定性较好,测试过程中不出现热分解、升华、化学反应等现象。
对药物制剂中晶型定量时,由于样品受到辅料的稀释,样品峰的总强度减弱,所要求的检测限提高。此外,由于操作或仪器的偏差及样品制备时不可避免地引入无机物等原因,使待分析物与基体之间的质量吸收系数的差异变大。为了减小以上因素的影响,制剂中晶型的定量通常采用多种方法结合或利用化学计量学方法。比如是Varasteh等利用XRPD与IR及Raman相结合,成功地定量了由 ALZA 公司开发的 片剂中 REJ-333369B晶型的含量,其检测限在2 %左右,体现了较好的灵敏度。又如Tong等利用XRPD、DSC和HPLC联合定量了沙美特罗昔萘酸酯粉末的相对含量。方法验证结果表明,该法准确度较高、重现性良好。此外,Lefort 等利用ss-NMR和DSC 联合定量了海藻糖制剂中的无定型的含量。
片剂中 REJ-333369B晶型的含量,其检测限在2 %左右,体现了较好的灵敏度。又如Tong等利用XRPD、DSC和HPLC联合定量了沙美特罗昔萘酸酯粉末的相对含量。方法验证结果表明,该法准确度较高、重现性良好。此外,Lefort 等利用ss-NMR和DSC 联合定量了海藻糖制剂中的无定型的含量。
总结及展望
药物晶型定量方法多种多样。对于特定的多晶型体系,有时可能很难判断哪种方法更好, 因此选择一种合适的定量方法需综合考虑方法的难易程度、灵敏度、耐用性、准确度和待分析试样特性等方面,并通过大量的实验考察和对比从而得出最优解。以上提到的定量方法可以单独或结合使用,对于制剂而言,由于辅料可能造成的干扰,往往考虑多种方法结合使用。
目前,XRPD仍是广大研究者们进行晶型定量分析的首选方法,但必须考虑到晶体的择优取向与试样制备过程晶型转变对定量分析的准确度造成的影响。因此,需通过合适的方法和手段来进一步提高该方法的准确度和可靠性。随着各种软件的出现及更新,全谱图法将是未来发展的趋势之一;光谱法中,随着Raman和NIRS灵敏度的提高以及 NIRS无需样品制备、专属性好等优势的发挥,未来光谱法在定量方面的应用会日益增多;热分析仪器精度及加热速率范围的扩大,使得此法能将不同晶型的特征峰分开,从而扩大了其在晶型定量方面的应用,但是其加热过程造成样品的不可逆破坏,在一定程度上限制其应用;ss-NMR 由于成本太高,其在固态药物分子水平分析的潜力尚未完全发挥。随着仪器技术水平的不断提升,未来还会有更多的联合分析技术得到应用。
总而言之,为确保同种药物中不同晶型对药物的质量和安全不会产生不良影响,药物晶型的定量分析方法开发,在今后的药物研发和生产过程中仍是一个不可或缺的研究热点。
参考文献
[1] 李钢,徐群,李瑞,等。那格列奈的多晶型与溶解度[J]. 化学学报,2007,65(24): 2817-2820.
[2] 李钢,徐群,莫祥银,等。不同晶型那格列奈的稳定性研究[J]. 化学学报,2003,61(2): 291-294.
[3] 周新波,吴素香,孙梦莹,等。埃索美拉唑镁盐多晶型的X-射线粉末衍射定量分析[J].药物分析杂志,2015, 35(11): 1934-1939.
[4] 徐婷,王彦,朱美兰,等。盐酸文拉法辛多晶型X-射线粉末衍射定量分析[J].。中国新药杂志,2017, 26(21): 2614-2619.
[5] Tiwari M, Chawla G, Bansal AK. Quantification of olanzapine polymorphs using powder X-ray diffraction technique [J]. J Pharm Biomed Anal, 2007, 43: 865−872.
[6] Moore MD, Cogdill RP, Wildfong PLD. Evaluation of chemometric algorithms in quantitative X-ray powder diffraction (XRPD) of intact multi-component consolidated samples [J]. J Pharm Biomed Anal, 2009, 49: 619-626.
[7] Croker D M, Hennigan M C, Maher A, et al. A Comparative Study of the Use of Powder X-Ray Diffraction, Raman and Near Infrared Spectroscopy for Quantification of Binary Polymorphic Mixtures of Piracetam [J]. J Pharm Biomed Anal, 2012, 63: 80-86.
[8] Uvarov V, Popov I. Development and metrological characterization of quantitative X-ray diffraction phase analysis for the mixtures of clopidogrel bisulphate polymorphs [J]. J Pharm Biomed Anal, 2008, 46: 676-682.
[9] 马乐伟,杜葳,赵春顺。药物晶型定量分析方法的研究进展[J]。药学学报,2011, 46(8): 896-903.
[10] Nemet Z, Sajo I, Demeter A. Rietveld Refinement in the Routine Quantitative Analysis of Famotidine Polymorphs [J]. J Pharm Biomed Anal, 2010, 51(3): 572-576.
[11] Sheikhzadeh M, Rohani S, Jutan A, et al. Quantitative and Molecular Analysis of Buspirone Hydrochloride Polymorphs [J]. J Pharm Sci, 2007, 96(3): 569-583.
[12] 杨 敏,徐 亮。采用差示扫描量热法定量分析固体分散体中的依折麦布晶型[J]。中国医药工业杂志,2023,54(3): 424-428.
[13] Guinot S, Leveiller F. The use of MTDSC to assess the amorphous phase content of a micronised drug substance [J]. Int J Pharm, 1999, 192: 63−75.
[14] Tong HHY, Shekunov BY, York P, et al. Thermal analysis of trace levels of polymorphic impurity in salmeterol xinafoate samples [J]. Pharm Res, 2003, 20: 1423−1429.
[15] Paul G, Paul C, Tim M, et al. A high-sensitivity, high-speed DSC technique: measurement of amorphous lactose [J]. Am Lab, 2003, 35(17): 17-22.
[16] McGregor C, Saunders MH, Buckton G, et al. The use of high-speed differential scanning calorimetry (Hyper-DSC (TM)) to study the thermal properties of carbamazepine polymorphs [J]. Thermochim Acta, 2004, 417: 231-237.
[17] Salari A, Young R E. Application of Attenuated Total Reflectance FTIR Spectroscopy to the Analysis of Mixtures of Pharmaceutical Polymorphs [J]. Int J Pharm,1998(163): 157-166.
[18] Braga J, Poppi R. Figures of merit for the determination of the polymorphic purity of carbamazepine by infrared spectroscopy and multivariate calibration [J]. J Pharm Sci, 2004, 93: 2124−2134.
[19] Kachrimanis K, Rontogianni M, Malamataris S. Simultaneous quantitative analysis of mebendazole polymorphs AC in powder mixtures by DRIFTS spectroscopy and ANN modeling [J]. J Pharm Biomed Anal, 2010, 51: 512−520.
[20] Patel AD, Luner PE, Kemper MS. Low-level determination of polymorph composition in physical mixtures by near-infrared reflectance spectroscopy [J]. J Pharm Sci, 2001, 90: 360-370.
[21] Blanco M, Villar A. Development and validation of a method for the polymorphic analysis of pharmaceutical preparations using near infrared spectroscopy [J]. J Pharm Sci, 2003, 92: 823-830.
[22] Blanco M, Valdés D, Bayod MS, et al. Characterization and analysis of polymorphs by near-infrared spectrometry [J]. Anal Chim Acta, 2004, 502: 221−227.
[23] Roberts S N C, Williams A C, Grimsey I M, et al. Quantitative Analysis of Mannitol Polymorphs: FT-Raman Spectroscopy [J]. J Pharm Biomed Anal, 2002(28): 1135-1147.
[24] Kachrimanis K,Braun D E,Griesser U J. Quantitative Analysis of Paracetamol Polymorphs in Powder Mixtures by FT-Raman Spectroscopy and PLS Regression[J]. J Pharm Biomed Anal, 2007, 43(2): 407-412.
[25] Vickery R D, Nemeth G A, Maurin M B. Solid-State Carbon NMR Characterization of the Polymorphs of Roxifiban[J]. J Pharm Biomed Anal, 2002, 30(1): 125-129.
[26] Tozuka Y, Ito A, Seki H, et al. Characterization and Quantitation of Clarithromycin Polymorphs by Powder X-Ray Diffractometry and Solid-State NMR Spectroscopy[J]. Chem Pharm Bull, 2002, 50(8): 1128-1130.
[27] Varasteh M, Deng Z, Hwang H, et al. Quantitative determination of polymorphic impurity by X-ray powder diffractometry in an OROS® formulation [J]. Int J Pharm, 2009, 366: 74-81.
[28] Tong HHY, Shekunov BY, Chan JP, et al. An improved thermoanalytical approach to quantifying trace levels of polymorphic impurity in drug powders [J]. Int J Pharm, 2005, 295: 191-199.
[29] Lefort R, De Gusseme A, Willart JF, et al. Solid state NMR and DSC methods for quantifying the amorphous content in solid dosage forms: an application to ball-milling of trehalose [J]. Int J Pharm, 2004, 280: 209-219.