随着目前医疗器械行业日新月异的高速发展,国内外的相关法规标准也对医疗器械生产环境也提出了更高的要求。关于医疗器械生产洁净室(区)的国内外法规一系列更新,特别是24年8月发布的新版YY/T 0033《无菌医疗器械生产洁净室 (区) 确认及监测要求》(征求意见稿)中,对无菌医疗器械生产洁净室(区)确认及监测提出了明确的要求。
核心术语须知
洁净室(区)clean room(zone)
需要对空气悬浮粒子及微生物含量进行控制的房间(区域)。其建筑结构、装备及作用均具有减少该房间(区域)内污染源的介入、产生和滞留的功能。
洁净度 cleanliness
洁净环境内单位体积空气中含大于或等于某一粒径的空气悬浮粒子和微生物的允许统计数。
洁净室(区)确认 clean room(zone)qualification
证实无菌医疗器械生产洁净室(区)是否能够满足规定要求的过程。
风险评估 risk assessment
识别出危险并对与暴露危险相关的风险进行分析和评价的系统性过程。
为无菌医疗器械生产洁净室(区)设定的某一参数(如微生物)的量值,超过该值,预示可能偏离正常状况,宜加强关注或采取纠正措施。
措施限 action level
为无菌医疗器械生产洁净室(区)设定的某一参数(如微生物)的量值,超过该值,表明已偏离正常状况,需立即查明原因并采取纠正措施)。
纠正措施 corrective action
当监测结果表明警戒限或措施限已被超过时,需要采取的措施。
医疗器械生产洁净室 (区)的洁净度等级应如何划分?
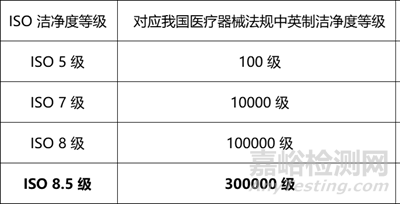
洁净度等级中的ISO 8.5 级洁净度等级从何而来?
需保留ISO 8.5 级对应的静态指标吗?
将采用国际通用的 ISO 等级(ISO 5/7/8/8.5 级),对应我国法规英制100/10000/100000/300000 级;
在医药行业所关注的≥0.5μm 粒子浓度,英制 300000 级洁净度需通过 ISO 等级公式计算,代入英制 300000 级对应的≥0.5μm 粒子浓度(约 11100000 个 /m³),反推得 ISO 等级约为 8.47,取近似值为8.5,故英制 300000 级对应 ISO 8.5 级;
值得注意的是,GB/T 25915.1-2021(等同 ISO 14644-1:2015)规定 ISO 8.5 级仅适用于动态,但我国医疗器械法规(如《医疗器械生产质量管理规范附录无菌医疗器械》)仍明确允许在英制 300000 级(对应 ISO 8.5 级)洁净室(区)进行部分无菌医疗器械生产,且该等级现行指标为静态指标,为适配国内法规要求及生产实际,新版YY/T 0033《无菌医疗器械生产洁净室 (区) 确认及监测要求》将 ISO 8.5 级对应的指标应用于静态。
无菌医疗器械生产洁净室(区)确认阶段/内容有哪些?
确认项目的方法包括哪些?如何选择?
首次确认:洁净室(区)投入使用前,需确认其洁净度等级符合要求。再确认:包括定期再确认(需定期开展,常用项目包括:空气悬浮粒子、气流方向、单向流风速/换气次数、高效过滤器完整性)、触发再确认(洁净室(区)长期停用后复启(新增),或设备、设施、工艺发生变更后,需视情况进行再确认);
无菌医疗器械生产洁净室(区)确认项目需覆盖物理参数、空气悬浮粒子、微生物三大核心维度。物理参数目前均按GB 50591-2010检测,但是国际标准ISO 14644-3也规定了洁净室(区)物理参数的测试方法,ISO 14644-3最新版已等同转化为我国国标GB/T 25915.3-2024。考虑到GB/T 25915.3中的方法为国际通用方法,可优先考虑采用GB/T 25915.3-2024中的检测方法。温湿度的检测暂不考虑采用GB/T 25915.3-2024中规定的方法,是因为 GB/T 25915.3 未对具体温湿度采样点进行规定,在使用时可能造成困惑,故仍要求按GB50591-2010进行检测。空气悬浮粒子参考GB/T 16292方法检测。空气微生物(沉降菌、浮游菌)参考GB/T 16293、GB/T 16294方法检测。表面微生物可参考《中国药典》四部 9205方法检测。
无菌医疗器械生产洁净室(区)的监测要求有哪些?
洁净室(区)监测计划为何要求“必须动态条件下监测”?
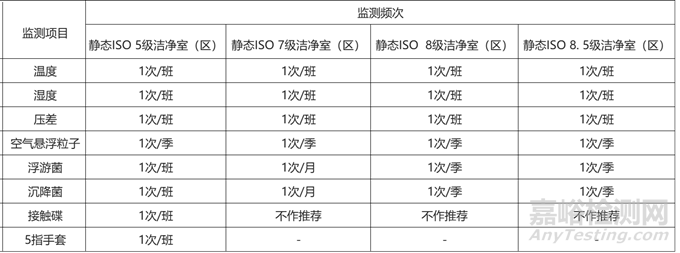
监测项目频率应如何计划安排?
监测项目及指标:常用项目为温度、相对湿度、压差、空气悬浮粒子、浮游菌、沉降菌、表面微生物等;
监测方法:需明确采样点位置、采样量、采样时间、微生物培养条件(如时间、温度、需氧和/或厌氧条件);
设备要求:明确监测设备的准确度及维护校准要求;
监测状态:必须在动态条件下进行监测;
监测频次:给出推荐频次,可基于风险评估调整;
限值设置:对空气悬浮粒子、微生物等关键项目设警戒限和措施限;
纠正措施:明确结果超限时的具体纠正措施;
数据管理:规定数据记录格式、趋势分析方法、记录存留要求;
计划审核:明确监测计划的审核频次。
动态监测的原因:动态条件指洁净室(区)处于 “正常生产状态”(含人员操作、设备运行、物料传输),此时的污染风险(如人员活动、设备运行产生的粒子、微生物)最接近实际生产场景;而静态条件(无人员、设备停运)无法反映真实生产中的洁净度变化。无菌医疗器械生产对环境洁净度的要求需贯穿全生产过程,动态监测能更准确评估实际生产环境是否符合要求,避免静态合格但动态超标的情况,故监测需在动态条件下进行。
洁净室(区)常用监测项目推荐的监测频次如下表。推荐的监测频次可基于风险评估及持续监测结果予以调整。