摘要
目的:总结我国儿童用药临床试验期间药物警戒监管体系现状与不足,以期为进一步提升我国儿童用药临床试验期间药物警戒监管工作能力提供参考。
方法:采用理论研究与比较分析法,阐述了我国临床试验期间药物警戒法规现状,总结了针对儿童用药临床试验期间药物警戒法规制定的必要性,对比了儿童用药与成人用药安全性报告审核思维的差异,对监管机构提出建议。
结果:我国现有临床试验期间药物警戒法规多为针对整体人群,缺乏儿童用药相关法规。儿童用药临床试验期间安全性报告审核思维与成人报告差异较大,应当在制定审核标准时予以考虑。结论 我国现有儿童用药临床试验期间药物警戒监管工作需进一步加强。
儿童用药品种短缺、剂量不明确是常见的共性问题,很重要的原因在于儿童用药研发技术要求高、临床试验风险大、收益率低等,造成企业研发积极性较差[1]。我国新药研发起步晚、基础薄弱,更加剧了这一问题。为了破解这一难题,我国出台了一系列鼓励儿童用研发措施,持续推进药品审评审批制度改革,完善药品审评审批体系[2]。改革成效显著,据统计,2022 年共有 66 个儿童用药获批上市,比 2021 年增加了 40.4%[3]。中国新药注册临床试验进展年度报告显示,2021 年登记的含儿童受试者的临床试验 168 项,比 2020 年增加了30.2%[4-5]。越来越多的儿童用药加速进入临床试验,如何能有效地保障儿童受试者的安全,是目前面临的一个重要问题。虽然在开展临床试验前,已获得了非临床试验数据、成人数据等支持开展临床试验的证据,但是儿童尚处于生长发育时期,肝肾功能、血脑屏障、神经系统等与成人差异较大,对药物的反应和成人可能会不同,简单地用成人数据外推可能会造成严重后果,比如氯霉素早期根据成人数据直接用于新生儿的感染,造成了患儿发生灰婴综合征死亡等[6]。所以加强儿童用药临床试验期间药物警戒监管工作显得尤为重要。
一、我国临床试验期间药物警戒法规
1.1 法规现状
我国加入人用药品技术要求国际协调理事会(ICH)后,临床试验期间药物警戒得到了快速发展,从 2018 年 5 月 ICH 临床安全数据管理指南(E2A)、个例安全性报告规范(E2B)的全面实施开始,到《药物警戒质量管理规范》(GVP)的发布,法规在不断完善[7]。现有临床试验期间药物警戒法规体系主要由 4 层框架构成:①法律层面,2019 年修订版《中华人民共和国药品管理法》《中华人民共和国疫苗管理法》[8-9]中,对申办者在临床试验期间安全性风险控制的责任进行了规定,并赋予了国家药品监管部门对临床试验安全性进行监督管理权限,从法律层面保证了研发期间药物警戒工作的开展;②行政法规方面,2022 年发布的《药品管理法实施条例》(修订草案征求意见稿)中[10],明确了申办者临床试验期间对药物安全性信息收集评估处置与报告等责任;③部门规章方面,2020 年 7 月 1 日起施行的《药品注册管理办法》中对申办者在临床试验期间发生的非预期严重不良反应和其他潜的严重2安全性风险信息的收集上报做了详细规定[11],国家药品监管部门可根据申办者提供的信息,评估临床试验安全性风险,采取必要措施,同时实施的配套文件《药物临床试验期安全信息评估与管理规范(试行)》中,从申请人的风险评估、药品监督管理部门风险评估与管理、工作程序、沟通交流等多方面,对药物临床试验期间安全信息评估与管理进行了全面阐述,具有非常强的可操作性;④规范方面,2020 年 7 月 1 日起施行的《药物临床试验质量管理规范》以及 2021 年 12 月 1 日起实施的《药物警戒质量管理规范》中[12-13],对申办者及临床试验机构、伦理委员会等各方在临床试验安全性评估管理中所承担的责任义务进行了详细规定,关注点由之前的严重不良事件转变为可疑且非预期严重不良反应,与 ICH E2A 相关要求一致,与国际接轨。
1.2 儿童用药临床试验期间药物警戒现状
根据前文所述,虽然目前我国对临床试验整体人群形成了以《中华人民共和国药品管理法》为主干,行政法规、部门规章相结合的多层次的监管法规体系,但缺乏针对特殊人群,尤其是儿童用药临床试验期间安全性监管法规,存在儿童用药药物警戒未能得到有效监管的隐患。我国 2018 年已充分实施 E2A,要求申请人按时限评估上报可疑且非预期重不良反应,但 E2A 仅仅是对行业的基本要求,是受试者保护的最后一道防线。儿童用药临床试验期间药物警戒是否仅参考整体人群即可,亦或是有哪些不同之处,亟需相关指导原则。
1.3 制定儿童用药临床试验期间药物警戒相关指导原则的必要性
儿童用药问题具有非常高的社会关注度,国家药品监督管理局药品审评中心(简称“药审中心”)采取了多项措施:①对于儿童用药采用优先审评审批政策,缩短审评用时,加快上市;②成立了儿童用药专项小组,负责儿科药物审评标准的建立,提高审评效率;③发布相关儿童用药指导原则,截至 2022 年底共发布了 13 项儿童用药指导原则,并在中心官网开通了“儿童用药专栏”,便于业界对儿童用药法规、动态的查询[14]。药审中心对儿童用药研发从政策到人力资源给予了充分的支持,也获得了非常好的效果,2022 年 60 余个儿童用药获批上市。但另外一方面,临床试验期间的药物警戒作为药物研发中的非常关键一环,承担着保护儿童受试者安全的重要作用,却在儿童用药指导原则中未得到相应重视,至今尚未有相关指导原则发布。在加快儿童用药审评审批的形势下,大量开展儿童用药临床试验,采用成人药物警戒指导原则及思维模式开展儿童用药临床试验期间药物警戒,存在一定的安全隐患,所以亟需相关指导原则指导儿童用药临床试验期间药物警戒活动。
二、我国临床试验期间药物警戒监管模式
2.1 临床试验期间安全性报告及监管措施的传递
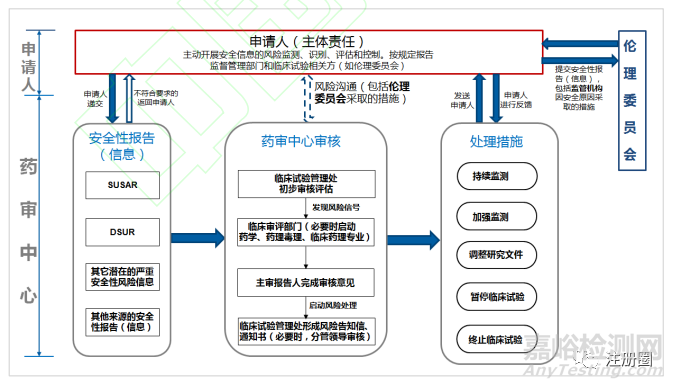
在临床试验期间,受试者安全的保护是重中之重,申办者、伦理委员会、药监机构各司其职,做到信息和审核处理意见的及时传递。申请人在获知发生严重不良事件时,要迅速进行分析评估,包括可预期性、药物的相关性、严重程度。判断为符合可疑且非预期严重不良反应(SUSAR)的,要按照 ICH E2A、E2B 进行快速向药监机构上报,同时按照《药物临床试验质量管理规范》(GCP)要求上报医院伦理委员会[15]。申请人还应定期进行汇总分析,对累积性安全信号进行识别评估,符合其他潜在安全性信息的,也应按 ICH E2A 要求进行上报[16]。另外按照 ICH E2F 要求,每年撰写定期安全性更新报告,对药物安全性进行总体评估。对于在临床试验中发现的安全性风险及时制定相关风险控制措施,对因安全性原因采取的重大措施,及时报药监机构及伦理委员会。伦理委员会对申请人提交的安全性信息及时进行审查,对发现可能存在安全性问题的应要求申请人采取相关措施,必要时可叫停临床试验[17]。对于伦理委员会的重要审核意见,申请人应及时在临床试验登记平台进行信息更新,并在研发期间定期安全性更新报告中进行记录总结。药审中心对申请人采取的措施进行评估,同时参考伦理委员会意见,如审核后认为受试者安全仍未能得到保障时,可要求申请人加强监测、修改方案、知情同意书、研究者手册等。申请人修改后反馈药审中心,同时报伦理委员会审核[18]。必要时,药审中心可暂停或终止临床试验,申请人收到暂停或终止临床试验通知书时,应立即采取行动,并及时递交伦理委员会(图 1)。
图1-临床试验期间药物警戒信息传递示意图
由图 1 可以看出,在申请人落实主体责任、伦理委员会发挥审查作用、药品监管部门履行监督义务的情况下,基本可以保证临床试验期间安全性报告和监管措施的及时传递,暂无需对儿童用药进行单独调整。未来结合中国疫苗国家监管体系(NRA)评估时,国外专家提出的药品监管部门和伦理委员会直接进行信息传递等建议,可考虑从儿童用药临床试验开始试点,更好地保护儿童受试者安全。
2.2 儿童用药临床试验期间安全性报告特点以及与成人用药审核思维的差异
2.2.1 报告数量方面差异较大
儿童用药临床试验期间 SUSAR 报告相对成人报告数量较少。以药审中心 2022 年收到的国内首次报告为例,其中 14 岁以下受试者 SUSAR 报告仅占全部报告的 1.38%。有限的数据对汇总分析提出了挑战,所以通常采用人工逐例审核的方式。
2.2.2 药物适应证方面差异较大
药审中心收到的报告主要以肿瘤药物为主,约占全部报告的 88.90%。但是在收到的儿童 SUSAR 报告中,却是以疫苗临床试验为主(占 74.60%),其次是肿瘤(占 6.35%)和系统性红斑狼疮(占 3.97%),显然在药物适应证方面存在较大差异。
2.2.3 安全性信号发现方面差异较大
疫苗临床试验基本为健康受试者,对安全性要求更高。肿瘤药物对安全性耐受程度相对较大,死亡事件经常作为试验的终点,所以肿瘤药物安全性信号通常是在大量数据累积的基础上发现的。而对于健康受试者,即使是严重性标准为住院,甚至其他重要医学事件时,即可产生出强烈的安全性信号[19]。比如 BC0335 颗粒,该品种适应证为儿童呼吸道合胞病毒感染,2021 年发生了因急性肝损伤导致住院的 SUSAR,经药审中心审核后,认为本品适用人群为儿童,耐受力较差,风险较大,发送了暂停临床试验通知书。
三、对儿童用药临床试验期间药物警戒监管工作的建议
3.1 制定相关指导原则
建议出台针对特殊用药人群(尤其是儿童)临床试验期间安全性监测评估相关指导原则。一方面,可参考国外的先进经验和上市后药物警戒经验,虽然临床试验期间药物警戒与上市后药物警戒存在一定差异,但很多方法、理念是可以借鉴的[20]。美国对于儿科用药相关法规制定发布较早,比如非常具有影响力且现在仍适用的两部法律:《儿童最佳药品法》(Best Pharmaceuticals for Children Act,BPCA)和《儿科研究公平法》(Pediatric ResearchEquity Act,PREA)[21-22],对鼓励企业研发、加强儿科用药药物警戒进行了详细规定。另外,21 CFR Part 50 Subpart D 对临床研究中儿童的额外保障进行了要求[23]。欧盟 2006 年发布的《儿科药品管理条例》(Paediatric Regulation)(第 1901/2006 号(EC)指令)[24]。与美国类似,一方面从审批形式、专利保护等多举措鼓励企业研发,另外从伦理审查等角度5加强了儿童受试者保护。在欧盟 GVP 中,制定了《产品或人群特殊考虑因素 IV:儿科人群》,从儿童年龄、生长发育情况等方面详细阐述了儿童用药药物警戒考虑因素,并对药物警戒活动中的安全风险管理、不良反应报告、信号的检测、管理、沟通(包括与儿科委员会的沟通)进行了详细介绍[25]。另外,要根据中国现有的监管理念、经验,坚持独立自主思考和解决实际问题。虽然我国临床试验期间药物警戒起步较晚,但从 2018 年 5 月至今,积累了大约 5年的数据,有很多经验、案例可以与世界分享。药审中心也在积极探索数字化、智能化的药物警戒监管模式,为世界贡献更多中国智慧。所以在制定指导原则时,在借鉴国外监管机构法规的同时,更要保持独立自主。
3.2 修订相关审核标准
在修订药审中心内部临床试验期间安全性报告审核评估标准时,建议加入对特殊人群的考虑,尤其是儿童。目前,药审中心在审核安全性报告时,主要考虑因素包括:不良反应术语、严重性标准、相关性以及同品种信息等,虽然在实际审核过程中对于儿童受试者会给予关注,但未形成统一的标准,存在审核尺度不一致的风险。前文已阐述了药审中心收到的SUSAR 中,儿童和成人报告的不同点。可根据这些不同点,结合实际审核案例,制定儿童用药安全性报告审核要点,统一审核尺度,提高审核效率。
3.3 组建儿童用药药物警戒相关委员会
建议药审中心建立针对儿童用药的临床试验药物警戒安全委员会。美国食品药品监督管理局(FDA)通过儿科咨询委员会( Pediatric Advisory Committee,PAC)及公众参与等形式,提高审查的全面性、完整性和公开透明,最大限度地保护儿童受试者的安全[26]。欧盟也设立了儿科委员会(PDCO),提升监管专业能力。药审中心目前建立了儿童用药技术审评临床专家咨询委员会,主要为提高儿童用药审评工作的质量和效率而组建,以临床专家为主。而临床试验期间药物警戒经常涉及多学科,比如药理毒理学、药学、临床药理学、理学等。建议组建针对儿童用药的临床试验药物警戒安全委员会,研究解决儿童用药临床试中发生的重点难点问题,保障审核的科学公正。
综上,在儿童用药临床试验过程中,保护儿童受试者的安全至关重要。建议通过制相关指导原则和审核标准,建立相关委员会,不断提升我国儿童用药药物警戒监管力,以期为儿童受试者安全性提供有力的保障。
参考文献
[1] LIANG Q. Lack of motivation in clinical trials of the pediatric drug[N]. Economic information67daily(经济参考报), 2021-08-25(005).
[2] YUAN LJ, WANG LQ, WANG XY, et al. The role and consideration of acceleratedapprovalprocess in pediatric drug registration system[J]. Chinese Journal ofPharmaceuticals(中国医药工业杂志), 2022, 53(11): 1529-1538.
[3] LUO N. Review and approval of the pediatric drug are accelerated[N].ChinaPharmaceuticalNews(中国医药报), 2023-01-17(001).
[4] Center for Drug Evaluation, National Medical Products Administration. The Annual Report onProgress of Clinical Trials for New Drug Registration in China (2021)[EB/OL].(2022-06-07)[2023-03-16].
https://www.cde.org.cn/main/news/viewInfoCommon/1839a2c931e1ed43eb4cc049e189cb0.
[5] Center for Drug Evaluation, National Medical Products Administration. TheAnnual Report onProgress of Clinical Trials for New Drug Registration in China (2020)[EB/OL].(2021-11-10)[2023-03-16].
https://www.cde.org.cn/main/news/viewInfoCommon/d670723dd2f646722097b3cf005e052.
[6] PAN M. Analysis the Characteristic of the pediatric drug[J]. Journal ofPractical MedicalTechniques(实用医技杂志), 2013, 20(4): 403-404.
[7] NMPA.NMPA Issued the Announcement on Good Pharmacovigilance Practice (No.65 of2021)[EB/OL].(2021-05-07)[2023-06-01].
https://www.gov.cn/gongbao/content/2021/content_5629614.htm.
[8] Pharmaceutical Administration Law of the People's Republic ofChina[EB/OL]. (2019-08-26)[2023-06-01].https://www.gov.cn/xinwen/2019-08/26/content_5424780.htm.
[9] Vaccine Administration Law of the People's Republic of China[EB/OL].(2019-06-29)[2023-06-01] http://www.npc.gov.cn/npc/c30834/201907/11447c85e05840b9b12c62b5b645fed.shtml.
[10] NMPA . NMPA publicly solicits opinions on the ImplementationRegulations ofPharmaceutical Administration Law of the People's Republic of China[EB/OL].(2022-05-09)[2023-0601]
https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20220509222233134.html.
[11] State Administration for Market Regulation.Provisions for DrugRegistration[EB/OL]. (2020-01-22)[2023-06-01]
https://www.samr.gov.cn/zw/zfxxgk/fdzdgknr/fgs/art/2023/art_3275cb2a929d4c4ac8c0421b2a9c257.html.
[12] National Medical Products Administration, National Health Commission. NMPA and NHCIssued the Announcement on Good Clinical Practice (No.57 of 2020)[EB/OL](2020-04-23)[2023-06-01].https://www.gov.cn/gongbao/content/2020/content_5525106.htm.
[13] NMPA.NMPA Issued the Announcement on Good PharmacovigilancePractice (No.65 of2021)[EB/OL].(2021-05-07)[2023-06-01]
https://www.gov.cn/gongbao/content/2021/content_5629614.htm.
[14] Center for Drug Evaluation, National Medical Products Administration.speed up the review and approval for the pediatric drug [EB/OL].(2023-01-17)[2023-06-01].https://www.cde.org.cn/main/news/viewInfoCommon/0fa129a8ad32929943ac11ef438959
30.
[15] National Medical Products Administration, National Health Commission. NMPA and NHC
Issued the Announcement on Good Clinical Practice (No.57 of 2020)[EB/OL]. (2020-04-23)[2023-
06-01]. https://www.gov.cn/gongbao/content/2020/content_5525106.htm.
[16] FENG HY, ZHOU LY, LI H, et al. Research III on Pharmacovigilance and Risk Control During 8
Clinical Trials of New Drugs: On Regulatory Requirements for Individual Case Safety Reports in
Clinical Trials of Drugs[J]. Chinese Pharmaceutical Affairs(中国药事), 2022, 36(6): 630-636.
[17] National Medical Products Administration, National Health Commission. NMPA and NHC
Issued the Announcement on Good Clinical Practice (No.57 of 2020)[EB/OL].(2020-04-23) [2023-
06-01]. https://www.gov.cn/gongbao/content/2020/content_5525106.htm.
[18] Center for Drug Evaluation, National Medical Products Administration. CDE issued the
Announcement on safety information evaluation and management practices during drug clinical
trials (No.5 of 2020) [EB/OL]. (2020-07-01)[2023-03-16].
https://www.cde.org.cn/main/news/viewInfoCommon/a1d42f512a341bc079ffb79df91f9cc7.
[19] CIOMS. Management of Safety Information from Clinical Trials(临床试验安全信息管理)
[M]//WANG HX, PEI XJ, CHEN J. Tianjin: Tianjin Science and Technology Translation and
Publishing Co., Ltd,2022.
[20] LI XX, TANG ZM, ZHOU J, et al. Limitations of the safety assessment in premarketing clinical
trials of drugs and suggestions for improvement[J]. Chinese Journal of Pharmacovigilance(中国
药物警戒), 2020, 164(8): 465-470.
[21] FDA. Best Pharmaceuticals for Children Act[EB/OL]. (2018-09-25)[2023-06-01].
https://www.fda.gov/drugs/development-resources/best-pharmaceuticals-children-act-bpca.
[22] FDA. Pediatric Research Equity Act[EB/OL]. (2019-11-07)[2023-06-01].
https://www.fda.gov/drugs/development-resources/pediatric-research-equity-act-prea.
[23] The U.S. National Archives and Records Administration. 21 CFR Part 50 Subpart D -Additional Safeguards for Children in Clinical Investigations [EB/OL]. (2001-04-24) [2023-03-16].
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-50/subpart-D.
[24] European Medicines Agency. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on Medicinal Products for Paediatric Use and Amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC)No 726/ 2004. [EB/OL]. (2006-12-27) [2023-03-16].
https://eurlex.europa.eu/legalcontent/EN/TXT/PDF/uri=CELEX:32006R1901&qid=1661481204018&from=EN.
[25] European Medicines Agency. Guideline on good pharmacovigilance practices (GVP) Productor Population-Specific Considerations IV: Paediatric population [EB/OL]. (2018-11-08) [2023-03-16].https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-goodphar.macovigilance-practices-gvp-product-population-specific-considerations-iv_en-0.pdf.
[26] FDA. Process for Handling Referrals to FDA Under 21 CFR 50.54 - Additional Safeguards for
Children in Clinical Investigations[EB/OL].[2023-03-16].
https://www.fda.gov/media/75222/download.