您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-09-19 22:36
杂质研究与控制是一项系统工程,CTD(CommonTechnical Document)申报格式体现了过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,更加符合杂质研究与控制的基本规律和逻辑思路。
Part1原料药CTD申报资料的相关要求
以原料药申报资料为例,CTD格式化申报资料中涉及杂质对比研究与控制的相关内容分布在(3.2.S.2)生产信息中的3.2.S.2.3物料控制,3.2.S.2.4关键步骤和中间体的控制;(3.2.S.3)特性鉴定中的3.2.S.3.2杂质谱分析;(3.2.S.4)原料药的质量控制中的3.2.S.4.1质量标准、3.2.S.4.2分析方法、3.2.S.4.3分析方法的验证、3.2.S.4.4批检验报告和3.2.S.4.5质量标准制定依据等各模块中,申报资料撰写中常犯的错误是人为割裂各项研究内容的相互联系,甚至遗漏相关研究内容,应高度关注杂质分析与控制的系统性与整体性,将杂质研究与控制的全部内容和信息体现在相应模块中。
Part2原料药工艺相关杂质控制策略
原料药工艺杂质的控制有以下4种方式。
方式一、在原料药质量标准中包含对杂质的检测,使用合适的分析规程将可接受标准设定在可接受限度以内。
举例:
根据各国药典收载情况及工艺合成路线,拟定将A-F杂质定入成品质量标准,根据代表性批次样品检测情况及稳定性样品检测情况结合各国药典的相关限度要求,制定该品种的放行标准及货架期标准。
方式二、在原料、起始物料或中间体的质量标准中包括对杂质的检测,或作为中控检测,同时制订可接受标准,或采用适当的分析方法,将杂质控制在可接受限度以下。
举例:
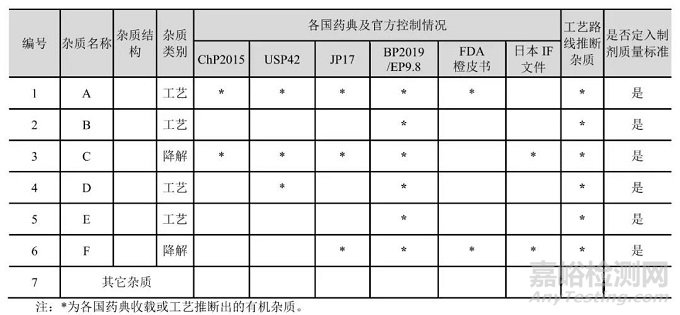
根据起始物料的合成路线及厂家的质量标准,该起始物料引入的杂质均可控制到限度以下,并在起始物料质量标准中进行控制,后续步骤将不再研究相关杂质。
方式三、在原料、起始物料或中间体的质量标准中包含对杂质的检测,或作为过程控制,制订一个高于原料药中杂质可接受限度的可接受标准,使用合适的分析规程并结合对杂质去向和被清除的理解,及相关的工艺控制,保证原料药中的杂质水平低于可接受限度而无需在后续工艺中再行检测。
举例:
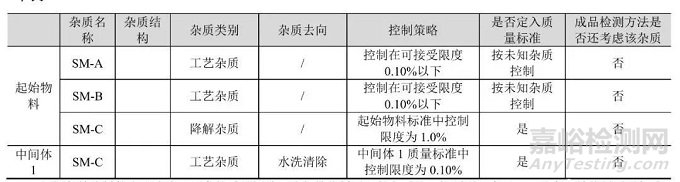
案例1
差2步到原料药时,生成了中间体X,杂质A在中间体X中常规检出。杂质A是一个工艺杂质,有可能带入到原料药。在实验研究中,将杂质A以不同浓度加入到中间体X中,发现即使在1%水平时,也能降到规定限度以下。由于中间体X的形成离原料药只有2步,杂质A在中间体X中的水平相对较高,另外又通过检测不同中试规模批次原料药里的杂质A水平,最终确认了工艺清除水平,获得的结果为可控制到限度以下。因此,对中间体X中杂质A控制在1.0%水平是可以接受的,不需要在原料药标准中对该杂质进行检测。
案例2
一个5步合成工艺中,起始物料Y在第3步引入,采用标准分析方法在起始物料Y中检出杂质B通常低于1.5%。为了确定1.5%的标准是否可接受,在实验室里进行了清除研究。将杂质B以不同浓度(最高10%)加入起始物料Y中,通过最后3步工艺步骤,得到清除系数>5000倍。该清除系数应用于起始物料Y含杂质B为1.5%时,杂质B在原料药中的期望水平远低于可接受标准(具有数量级的显著差异)。因此认为起始物料Y中杂质B为1.5%的质量标准是可接受的,不需要再提交中试生产数据或商业放大批数据。
方式四、了解工艺参数及其对残留杂质水平(包括去向和清除知识)的影响,确信原料药中的杂质水平将会低于可接受限度,则建议不需要对该杂质进行分析检测(即,不需要将杂质列在任何质量标准中)。
方法 4 特别适用于那些自身不稳定的杂质(例如,与水迅速完全反应的二氯亚砜),或那些在合成路线早期引入并可被有效清除的杂质。有些情况下,如果已经知道杂质如何形成或者是合成后期引入的杂质也可以采用方法4;但此时需要提交工艺特异性数据来论述该方法的合理性。
案例1
药典收载杂质,但本工艺路线明显不可能产生的杂质,可无需研究。
案例2
某起始物料在步骤2的纯化过程中能有效除去,在粗品步骤检测该起始物料的残留。并对中试样品进行了该起始物料残留检测,结果表明3批中试粗品样品中,该起始物料残留远低于可接受标准限度,该起始物料的结构类似杂质与其具有类似的反应活性,在该起始物料中控制限度为0.50%,根据3批中试粗品中该起始物料的残留情况,推算出粗品中该起始物料的结构类似杂质的量也远低于规定限度(具有数量级差别),且这些杂质在存放过程中不会产生,因此不必在成品中对该起始物料及类似杂质进行常规测试。
案例3
高活性杂质,如氯化亚砜遇水即发生反应,而合成过程中使用大量的水进行冲洗,因此不再提交任何实验室或中试数据。
案例4
某CDE发补资料要求补充研究三氟甲磺酸残留,经理论分析,该物质遇水易水解生成三氟甲烷和硫酸,因此标准中进行硫酸盐或三氟甲烷的检测即可。
控制方法的注意事项:
与采用方式3时一样,采用方式4时,如果仅仅根据科学原理来进行论述是不够充分的,强烈建议提交分析数据来支持控制方法。提交的资料可以适当包括在下游化学反应中导致杂质结构改变的信息(去向);中试批次的分析数据;某些情况下,可以包括实验室规模中有意加入杂质的研究(加标研究)。在这些情况下,一定要证明该杂质的去向/清除是可靠的,能够持续地保证最终原料药中杂质残留量超过可接受限度的可能性可以忽略。如果清除因子是根据研发数据得到的,一定要处理预期的规模依赖性或非依赖性。如果用于研发阶段的小规模模型被认为不能代表商业规模,则一般需要确认中试规模和/或初始商业批次中所用的控制是适当的。清除因子(由实验室或中试规模数据计算而来)数量级、杂质引入点和对下游工艺清除点的理解,决定了是否需要中试/商业批次数据。如果方式3和4不能得到合理论证,则在原料、起始物料或中间体的质量标准中应包括对杂质的检测,或作为过程控制(方式2),达到可接受限度或在原料药的质量标准中规定对杂质的检测,达到可接受限度(方式1)。对于在较后合成步骤中引入的杂质,除另有论述外,一般应采用方式1的控制方法。
Part3原料药降解产物的控制
对于降解产物一般要了解其降解途径与原料药和制剂的生产工艺和/或其拟定的包装和存贮条件的相关性。建议采用一个设计良好的加速试验(例如,40℃/75%相对湿度,6个月),采用拟定的包装形式,采用适当的分析方法来确定潜在降解产物的相关性。也可以采用一个设计良好的动力学等效的时间更短温度更高的稳定性试验,使用拟定的商业包装,在开始长期稳定性试验前确定降解途径的相关性。对于那些根据潜在降解途径的知识了解到的但在产品中尚未观察到的潜在降解产物,这类研究对了解其相关性特别有用。根据这些加速试验的结果,如果预计降解产物将在拟定包装和存贮条件下形成,且接近可接受限度水平,则应采取措施控制降解产物的形成。这种情况下,除另有论述外,应监控拟定存贮条件下(拟定的商业包装)长期稳定性试验主要时间点上原料药或制剂中的降解。降解产物的质量标准限度是否恰当通常取决于这些稳定性研究的结果。如果预计制剂研发和包装设计的选择不能将致突变降解产物水平控制在可接受限度以下,而杂质是“最低合理可行”水平,则可以根据风险/利益分析来论证更高限度的合理性。
个人理解,根据《中国药典》2015年版(四部)通则9001原料药物与制剂稳定性试验指导原则,稳定性试验包括影响因素、加速试验与长期试验。对于未知降解杂质的研究,影响因素条件超出鉴定限,就要引起关注,但不一定必须定性;加速稳定性试验(或动力等效研究,可按每提高10℃,时间减半评估)应基于影响因素考察选择适当的包材进行,若仍超鉴定限,建议进行定性研究;长期稳定性研究,若超鉴定限,必须进行定性研究。对于仿制药,如产品较为稳定,质量标准各项均不比各国药典差,不强求与参比制剂对比研究,基于前期研究对产品性质的了解以及允许的限度综合判断相应的风险,如超过预期的限度的风险较高,则应同时对参比样品进行对比研究,不得比参比样品差,最好为原研厂生产的原料药,如不能获得原研厂生产的原料药,可提供与原研制剂的杂质谱比较研究资料作参考。建议工艺确定后的小试样品就进行影响因素、加速放样,尽早发现降解杂质的规律,降低研发风险。
Part4原料药质量标准的建立
质量标准的定义
质量标准由一系列的检测项目、相应的分析方法和合理的可接受标准组成,这些可接受标准以限度值、范围或其他描述来表示。质量标准是重要的质量指标,它由生产商提出和论证,由监管机构批准并作为批准产品的依据。
质量标准的论证
首次提出质量标准时,应对每一个检测方法和每一个可接受标准进行论证。论证包括有关的研究开发数据、药典标准、用于毒理和临床研究的原料药及制剂的检测数据、加速试验和长期稳定性研究的结果;另外,还应考虑分析方法和生产可能波动的合理范围。全盘考虑是非常重要的。制订和论证质量标准时,应考虑稳定性批次和生产规模放大/验证批次的检测结果,特别是申报稳定性批次的结果。以有关物质限度的制定为例,主要从安全性角度考虑,如根据多批次研究结果显示杂质实测结果低于杂质安全性允许的最高限度,可降低限度,分析方法与生产可能带入的波动,应该做限度的减法。
个人理解
在进行质量标准制定时,1、应进行文献检测,包括但不限于各国质量标准、进口注册标准、各国官方网站的审评资料(如日本的IF文件等)、毒理和临床的相关期刊文献等。2、应对加速、长期稳定性研究数据进行汇总,3、充分考虑分析方法测定数据的波动及生产的可能波动,分析方法测定数据的波动可以以方法的回收率大致评估,严格来讲测定数据的波动(即方法的误差)应该是在一定置信水平上的波动范围,生产的可能波动可以以多批次测定结果的均值+3δ表示。
Part5原料药对制剂质量标准的影响
通常,原料药中已控制的仅和原料药质量相关的检测项目,在药物制剂中可不必重复检测,如在制剂中就不必检测已在原料药中控制过的非降解产物的工艺杂质。
新原料药降解产生的有机杂质和该制剂在生产过程中产生的杂质均应在新药制剂中监测。应对单个特定降解产物(包括已鉴定的和未鉴定的)及总降解产物的可接受限度进行规定。新原料药合成中生成的杂质(工艺杂质)通常在原料药的检测中已控制,因此不包括在制剂总杂质限度中。但当工艺杂质同时也是降解物时,应监测其含量并列入到总降解产物的限度中。当通过适当的分析方法最后证明原料药在新药申报中提出的指定处方和贮藏条件下不降解,经监管机构批准可以减免对降解产物的测定[2]。
对上述两段的个人理解为,原料药中已控制的工艺杂质,在制剂分析方法开发时,仅需对其进行定位,目的是不影响降解产物的检测,关于工艺杂质之间的分离度可不做要求,也不计入总杂。但《中国药典》2015年版(四部)通则9102中规定“除降解产物和毒性杂质外,在原料药中已控制的杂质,在制剂中一般不再控制”,什么属于毒性杂质?个人认为应基于杂质的TD50\LD50等数据进行评估,但对于仿制药,各国药典有限度要求的一般工艺杂质,在制剂质量标准建立中可不计入制剂总杂质统计。对于基因毒警示结构杂质,建议制剂在制定质量标准时需对其进行评估,若同时为降解杂质,建议定制质量标准。
Part6杂质研究分析方法的建立
对于仿制药杂质研究的分析方法应基于各国药典的基础上开展,所开发方法不得比药典方法差,建议以列表形式整理:
表1 有关物质方法开发对比1
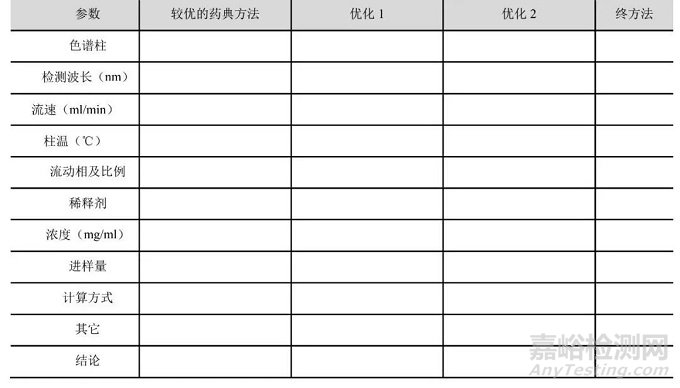
表 2有关物质方法开发对比2
分析方法确定过程应展现开发思路、摸索过程及细节钻研,与药典方法对比应明确优缺点,体现差异性。
举例1
某项目USP药典方法采用流动相作为稀释剂,ChP药典方法采用水做稀释剂。以水做稀释剂,杂质A的线性截距约为21%,回收率偏高,均值在120%以上。以流动相为稀释剂,杂质A的线性截距约为2%,回收率均值在90%-110%之间。
本例说明稀释剂的选择至关重要,溶剂效应不仅可能影响峰形,还有可能影响方法的准确度。
举例2
某项目采用溶液颜色对相关杂质进行控制,中国药典采用标准比色法,欧洲药典和日本药局方采用UV吸光度法。经分析,本品有色杂质可能不止固定的一种,其色调有时会随不同杂质而改变,甚至会由几种杂质混合贡献并随杂质组成的不同而变化,所以溶液的色调不固定,溶液的最大吸收波长也有漂移的可能。单一固定波长的UV法适用于固定单一显色杂质的控制,如用于本品则顾此失彼,有一定局限性。ChP采用与标准比色液比较的方法(包括目视与色差计法)可以较为灵活全面地监控有色杂质总量的变化,缺点量化性能差;JP对操作时间有限制提示,说明该溶液的稳定性可能不太好,因此对该溶液的稳定性进行考察,最终确定的方法为标准比色法,并规定了测定时间,整个研究过程可概括为“集齐——读懂—完善—超越”。
本例说明方法建立过程应站在前人研究的基础上进一步深化研究,知其然,知其所以然,做到透彻分析,有理有据。
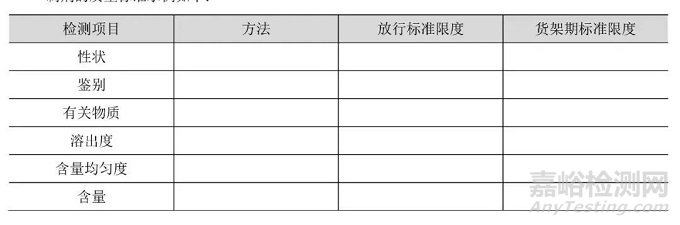
Part7货架期标准与放行标准
对于药物制剂,放行与货架期可接受标准可以不同,通常放行标准应严于货架期标准,如对含量和杂质(降解产物)的限度要求。在日本和美国,这一概念只用于内控标准,而不是法定放行标准。因此在这些地区,法定的可接受标准从出厂到货架期均相同,但申报者可选用更严格的内控标准作为放行依据,以确保产品在货架期期间仍符合法定的可接受标准。在欧盟,当放行和货架期标准不同时,管理机构要求提供各自的质量标准。
制剂的质量标准示例如下:
参考文献
[1] ICHQ6A
[2] ICHM7
[3]《中国药典》2015年版(四部)
[4] CTD格式申报资料解读与建议—余立

来源:Internet


