新版 ISO 10993-1 即将正式发布,当前处于 FDIS(最终草案国际标准)阶段,预计 2025 年 9 月会成为正式标准。修订不是颠覆性的,而是优化和细化,但对风险管理和材料评估提出了更高要求。
同时2025年8月21日,中检院发布了《医疗器械生物学评价 第1部分:风险管理过程中生物学安全性评价的要求和通用原则》,即GB/T 16886.1-2025征求意见稿,等同采用国际标准ISO/FDIS 10993-1:2025。
根据往年情况,国家药监局一向都是等ISO标准发布后再更新GB/YY标准,而此次紧跟ISO节奏,官网显示ISO 10993-1于2025年7月18日进入Under Publication阶段,预计于2025年10月5日前ISO 10993-1:2025会正式对外发布。而GB/T 16886.1根据其国家标准项目申报书中所示,预计于2026年12月报批。
新版标准条款解读
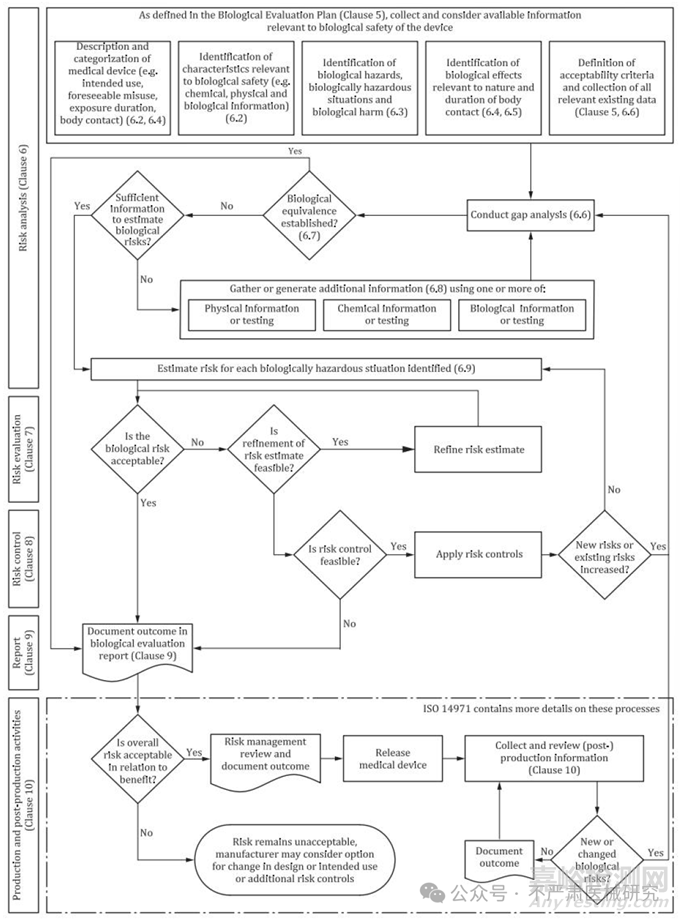
1. 风险管理与 ISO 14971 的深度融合
新版定位:“风险管理中的生物学评价原则”。
要求企业不再单纯依赖测试清单,而是基于 风险识别 → 风险分析 → 风险控制 → 风险可接受性论证。
具体表现:
必须在 生物学评价计划(BEP) 中明确说明风险分析逻辑。
生物学评价报告(BER)需要体现风险与数据的对应关系。
强调产品实际临床情境,而不是“一刀切”的测试。
2. 合理可预见的误用(Reasonably Foreseeable Misuse)
新增要求:必须考虑 合理可预见的误用。
举例:
导管本应短期使用,但可能被临床人员延长使用。
可重复使用器械在未按说明书正确清洗时继续使用。
要求:
对误用情况的暴露时间、方式进行风险评估。
必要时增加额外论证或补充测试。
3. 曝露时间的新定义:Contact Day
旧版:短期(≤24h)、长期(≤30天)、持久(>30天)。
新版:用 天数 更精细计算,称为 Contact Day。
日常接触是指医疗器械每天与人体接触任意时长的情况。对于这类医疗器械,总暴露期是指从首次使用该医疗器械,到对同一患者最后一次使用该器械或更换同类型器械之间的日历天数。
间歇接触是指使用器械时,连续两次接触组织之间至少间隔24小时。这可以是同一医疗器械的重复使用,或考虑更换同一医疗器械的情况。对于这些医疗器械,总暴露时间是通过累加同一患者从首次使用该医疗器械到最后一次使用的接触天数来计算的。
影响:计算接触时间更精细,更严苛,这可能会导致部分“边界型”器械的分类发生变化。
4. 生命周期(Life Cycle)考量
新版更强调全生命周期:
从生产、运输、储存、临床使用、再处理、直至废弃。
要求:需要评估 器械在不同阶段 的潜在风险。
例如:
可重复使用器械 → 清洗消毒后的残留风险;植入物 → 长期降解产物的毒性;
一次性器械 → 老化后的化学物质释放风险。
对材料的要求更严格,尤其是:
纳米材料(需考虑纳米级别颗粒释放及长期风险);
CMR 物质(致癌、致突变、生殖毒性);
ED 物质(内分泌干扰物);
要求更详细的化学表征报告(XPS、FTIR、GC-MS、ICP-MS 等)。
对比旧版,新版更强调 定量数据 和 毒理学阈值,减少“经验性”表述。
6. 动物试验的 3R 原则
明确提出:应 避免不必要的动物实验。
鼓励使用:现有数据(历史临床使用、文献、同类产品数据);
替代方法(体外模型、计算毒理学 QIVIVE)。
如果必须动物实验 → 需要说明理由,并遵循 Replacement, Reduction, Refinement。
7. 生物效应的识别
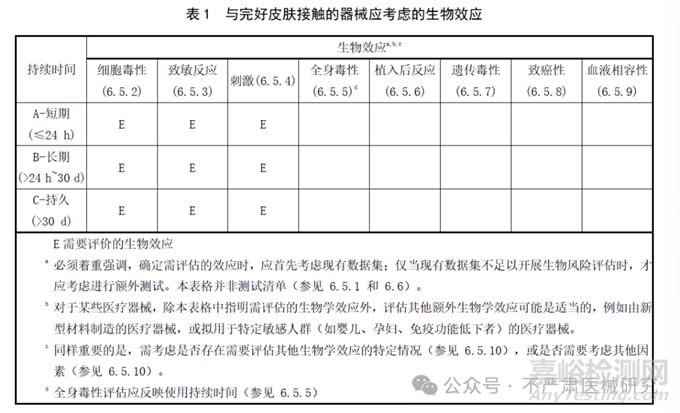
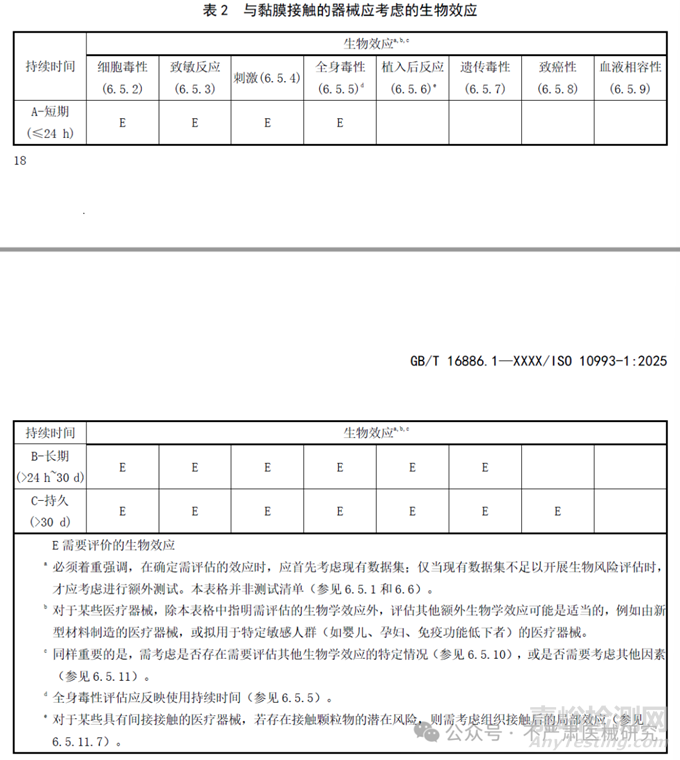
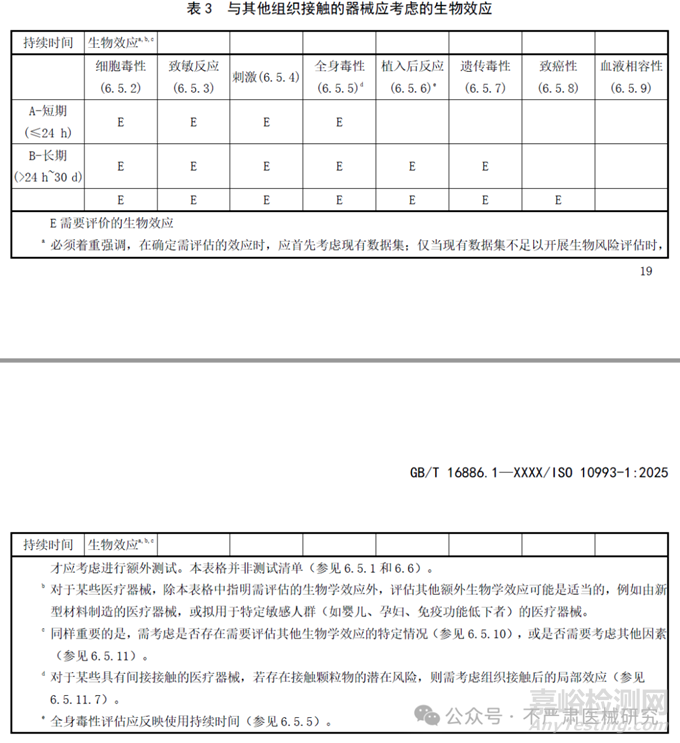
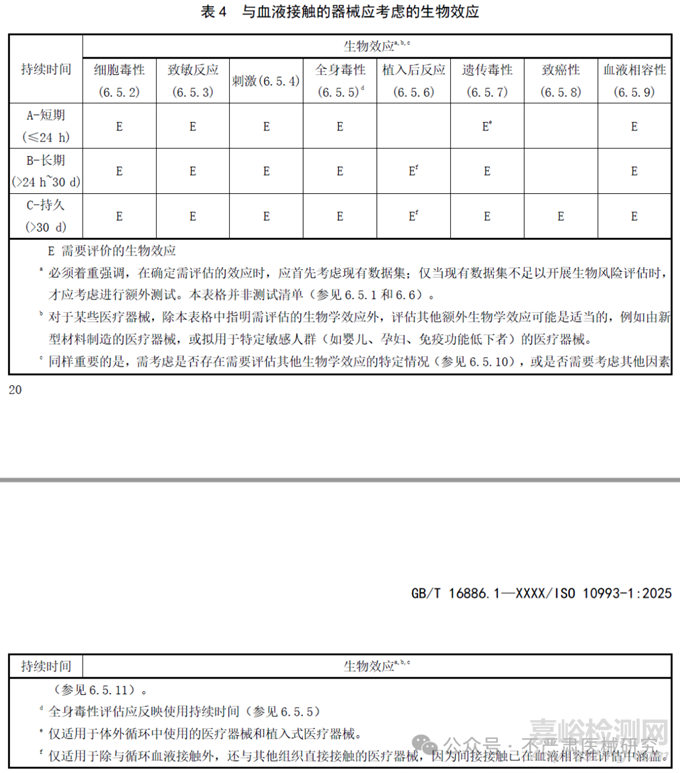
2018版的生物学评价终点矩阵(表A.1)被拆成4张新表(表1–4),每张表列出11个核心的“生物效应”:细胞毒性、致敏性、刺激性、全身毒性、植入反应、遗传毒性、致癌性、血液相容性,根据不同接触性质和接触时长分类,识别需评估的生物效应。
新4表差异:
完整粘膜B类增加了遗传毒性评估要求;
完整粘膜C类增加了致癌性评估要求;受损或破裂表面(皮肤或粘膜)B类增加了遗传毒性评估要求;
循环血液A类删除了局部效应(植入)评估要求;
急性全身毒性、亚急性全身毒性、亚慢性全身毒性、慢性全身毒性(综合为全身毒性)、组织接触后局部效应(更名自植入反应);
原生殖和发育毒性、材料介导的致热性分为“其他生物效应”。
8. 新增要求
以下为新增:
2个新增需要考虑的“生物效应”:
免疫毒性(新增):当材料的物理或化学特性、未知免疫原性或已有提示性数据表明可能引发异常免疫应答时需评估;
神经毒性(新增):当器械或其组分直接接触或间接接触中枢/外周神经组织或脑脊液时需评估;
8个“其他需考虑因素”:
与组织短暂接触:小于1min可书面豁免,重复用须累计暴露;若留涂层/润滑则必须详细评估;
低风险仅完整皮肤接触:仅在拿取中碰完好皮肤,材料有相同接触史且安全,需书面证明风险不适用;
经皮/黏膜吸收:乳膏、滴剂等若大量吸收须评估系统暴露并决定系统毒性试验;
发生变化的材料:设计在体内聚合/降解/吸收时,须逐阶段评价起始物、中间体、终产物及降解物;
材料降解:可吸收或磨损/腐蚀释放产物时,须记录产物、参数及降解机制;颗粒物:制造残留、降解或磨损所致颗粒须评估局部物理效应及溶出物全身影响;
毒代动力学:可吸收、长植、高释放、纳米或药械组合须评估吸收、分布、代谢和排泄(ADME)过程;若暴露已证安全可免;
有害成分:已知毒物且量可能有毒时,无论分类如何均须专项处理其风险。
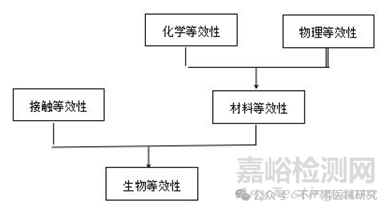
9. 新增生物等效性的评估路径:
若医疗器械在以下方面与另一款已有生物学安全性数据支持的医疗器械足够相似,则可证明该医疗器械具有生物学等效性,需要考虑以下方面:
——人体接触类型和接触时长;
——成分组成;
——与生物学安全性相关的物理特性或机械性能;
——制造工艺(包括包装和灭菌,如适用);
——患者人群;
——材料或医疗器械的尺寸或数量(数量或用量)。
如材料化学数据充分,可免做部分毒理实验;如临床等效性明确,可减少动物或人体试验。但要求透明记录论证逻辑,监管机构会重点审查。
实时时间对比
各国对于新版生物学标准实施时间与监管环境的对比:
|
ISO 10993时间线: |
GB/T 16886时间线: |
|
2025年5月:FDIS发布。 |
2026年1月:起草 |
|
2025年7月:投票截止。 |
2026年6~8月:征求意见 |
|
预计2025年9月正式发布。 |
2026年10月:审查 |
|
|
2026年12月:报批 |
|
影响范围:CE MDR → 会很快引用新版;FDA → 目前仍接受 2018 版,但预计会逐步过渡;中国 NMPA → 可能滞后 1–2 年采纳。
|
|
实际影响
对行业的影响:
1. 并不是要求所有产品重新做实验。
2. 更多是要求企业更新风险管理文件、生物学评价报告(BER),并确保论证逻辑符合新版要求。
3. 对于新产品、新材料,会面临更严格的合规路径。
对企业的实际影响
1. 文档更新
BEP/BER 必须更新,加入新版的风险逻辑、误用分析、Contact Day。
2. 测试计划调整
边界产品可能需要增加长期毒理学数据。
3. 材料供应链
对供应商的材料安全数据(特别是 CMR/ED/纳米)要求更严格。
4. 成本和时间
对新产品,准备工作量可能增加。
但通过减少冗余动物实验 → 可能在长期反而降低成本。