强制降解试验属于药品稳定性研究的内容之一,是研发分析人员在质量研究过程中无法绕开的一个话题。强制降解试验如能够合理、高效的开展,将为产品的研发以及生产检验提供极大的帮助。然而,查阅国内外相关的指导原则,除光降解试验外,其他强降解条件(如:高温、高湿、酸/碱水解、氧化等)均未进行明确规定。工作何时进行,如何开展,试验结果如何评估都困扰着不少研发分析人员。因此,一些初入职场的分析人员,一谈到强制降解试验,总是不知所措。
作为一名默默奋斗在基层的研发分析人员,通过不断学习以及工作中积累的经验,浅谈下个人对强制降解试验理解,分享给大家。
本文拟从以下4个方面谈一下这个话题:
强制降解试验的目的
何时开展强制降解试验
如何设计和开展强制降解试验
强制降解试验数据的评估
一、强制降解试验的目的
开展强制降解试验是获得药物可能的降解途径和降解产物信息的重要途径,以指导产品开发和分析方法开发。
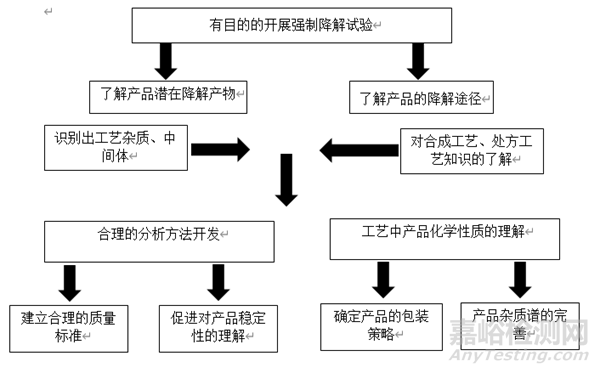
有助于更全面了解药物的降解情况及杂质谱概况。对分析方法的开发、质量标准的建立以及稳定性研究具有重要意义。也为药品的处方工艺开发、包装、贮藏条件的确定提供有益支持。图1展示了强制降解试验在产品质量研究过程中的重要作用。
▲图1-强制降解试验在产品质量研究过程中的重要作用。
二、何时开展强制降解试验
根据上面对强制降解试验目的的分析,我们可以在药物研发的各阶段根据项目研发的需求开展相应的强制降解试验。对于创新药,由于通过文献获取对化合物自身化学性质信息较少,对于此类药品,强制降解试验建议尽早开展,如产品开发阶段,分析方法建立阶段即可酌情开展相应工作,以对化合物性质有初步了解,指导产品开发和分析方法建立。随着研发的不断深入和对产品性质的了解加深,可通过设计比较完整的强制降解试验,对于全面了解药物性质及其稳定性,对产品处方工艺的确定,包装系统,以及储存条件的确定提供指导。对于仿制药,一般可通过文献调研了解参比制剂或同类药品的稳定特性及其降解途径,并结合药物结构和剂型特点等先验知识,有侧重地考察已有较明确提示信息的特定降解类型。特别是,当处方工艺或有关物质分析方法与参比存在较大差异时,可通过设计相应强制降解试验来确认/验证分析方法的专属性和灵敏度。也可通过采用特定破坏条件的样品,与另一实验室的检测结果进行比对,以评估分析方法在不同实验室的适用性。
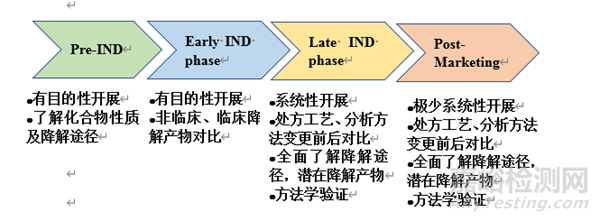
▲图2-何时开展相应的强制降解工作
图2展示了何时开展相应的强制降解工作。总结来说,Pre-IND和早期临床阶段一般有目的性开展相应的强制降解试验,以了解药物的化学性质及降解途径。临床阶段后期,因处方工艺、分析方法已确定,需设计系统性的开展工作,全面了解药物的降解途径,潜在降解产物及分析方法的适用性。药品上市后,一般在产品处方工艺或分析方法发生重大变更时候,设计强制降解试验,重新评估产品的降解产物以及分析方法的适用性。
三、如何设计和开展强制降解试验
由于不同药物品种化学性质不同,试验考察的目的不同,强制降解试验条件的选择千差万别,那么如何选择合适的降解条件,是广大分析研发人员需要考虑的事情。笔者查阅相关指导原则和文献,结合个人工作经验中的一些体会,对降解类型和试验条件的设置汇总如下表:
▲表1-降解类型和试验条件的设置
|
类型
|
破坏条件
|
破坏时间
|
|
酸破坏
|
常见0.1N~1N HCl溶液(可加温度)
|
破坏时间从几分钟到7天不等,破坏时间需要摸索(具体项目根据实际情况进行)。
|
|
碱破坏
|
常见0.1N~1N NaOH溶液(可加温度)
|
|
氧化破坏
|
0.1%~3%双氧水中性条件下室温破坏7天或20%双氧水破坏
|
|
高温破坏
|
固体
|
60~105℃
|
|
液体
|
40~80℃
|
|
光照破坏
|
固体
|
依据ICH Q1B进行(即总照度不低于1.2 × 106 Lux·h、近紫外能量不低于200w·h·m2)
|
|
液体
|
4500lx± 500lx,90uw/cm2条件下,照射一定时间
|
破坏样品一般溶解至适当溶剂中进行破坏,如药品水溶性较差,可加入适量有机溶剂进行溶解。初始条件可优先选择相对温和的条件下进行破坏,根据降解程度逐步调整破坏条件,降解程度一般控制在5%~20%,对于要求含量为标示量的90.0%~110.0%的药品,建议破坏程度不超过主成分的10%。避免破坏过度,造成二次降解。当然,由于药物自身的化学性质,并不是每个降解途径都能够达到目标降解程度,当药品在剧烈条件下破坏一定时间后,仍达到相应降解程度,则终止降解试验(如,1N HCl溶液室温破坏7天降解程度仍低于5%,可终止试验,不必追求破坏程度必需在5%~20%)。
四、强制降解试验数据的评估
强制降解试验完成后,应根据试验目的与药品特点,对试验结果进行分析和评估,并进行相应风险控制和改进优化。
4.1 药物降解途径分析和杂质谱研究方面
强制降解试验结束后,应立即检查样品物理性质(如外观、溶液的澄清度或颜色)的所有变化,并进行含量和降解产物的测定,通过结果分析充分理解试验条件对药物稳定性的影响,评估分析方法的适用性,为药物的处方工艺开发、包装策略的制定提供指导。
检测过程建议使用多种不同类型的检测器,如紫外检测器和质谱,以最大可能的发现药物潜在降解杂质。紫外检测器应使用二极管阵列检测器(DAD),以分析降解产物在不同检测波长下的吸收差异,使用质谱对强制降解样品进行分析,避免降解产生的一些无紫外吸收化合物无法检测。强制降解试验产生的主要降解杂质应根据常规稳定性研究结果分析及ICH Q3等指导原则相关要求,进行杂质鉴别和结构确认。
4.2 方法研究方面
主要的降解杂质与目标分析杂质及主成分之间应具有足够的基线分离度,且无干扰(溶剂峰和工艺杂质峰),根据实验结果及分析目标概况(ATP)对分析方法进行优化调整。
使用DAD检测器,并对主成分峰的峰纯度进行分析,以避免降解杂质峰与主成分峰的共洗脱现象。
应对降解后样品的质量平衡情况进行评估,可对降解后杂质的增加量与降解后含量之和,与初始的接近程度进行评估。一般要求降解后质量平衡在90%~110%,如出现较为严重的质量不平衡情况,应进行解释说明,并进行风险评估,对分析方法进行优化调整。
五、总结
设计合理且完善的强制降解实验,可以促进对药物降解知识的全面了解,也是分析方法开发的基础,方法学验证的重要内容,对质量标准的建立和稳定性研究都有重要作用。上述仅代表个人结合工作对强制降解试验的理解和认知,具体实施细节感兴趣的可参照下方文献进一步探讨。
参考文献
[1]浅谈化学药物强制降解试验的设计与开展[J]中国新药杂志,2019,28(20):2468-2472
[2] FDA perspectives: scientific considerations of forced degradation studies in ANDA submissions[J].Pharm Tech,2012,36( 5)
[3] Development of forced degradation and stability indicating studies of drugs-A review [J] Journal of pharm Analysis 2014(3)
[4] Forced degradation studies–comparison between ICH, EMA, FDA and WHO guidelines and ANVISA’s resolution RDC 53/2015 [D]
[5] ICH. Q1(R2)
[6] ICH. Q14 Draft Guideline