引言
《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》中明确指出了“应通过研究确定药液配制后至过滤前的存放时间、药液过滤操作的时间、过滤后至灌装前放置的时间等工艺时限来降低除菌过滤的风险。”
国内制药企业对此指导原则的解读大相径庭,导致了注射剂项目关于药液存放时限的考察在实际研发生产过程中的做法不一,本文章,以冻干粉针剂为例,参考CDE、EU及PDA相关指导原则并结合作者的研发生产经验,对注射剂项目研发生产过程中关于药液存放时限进行了系统性的考量,不足之处,还请业内同仁指出,共同学习。
一、国内外相关指导原则
1、国外相关指导原则
(1)EU GMP Annex 1 : Manufacture of Sterile Products(2020).
EU于2020-02-20发布GMP Annex 1 : Manufacture of Sterile Products(2020)修订草案关于药液存放时限的考量,如下:
从产品开始制备到通过除菌过滤器(如果适用)进行除菌或过滤直到无菌灌装过程结束之间的时间。考虑到产品的成分和规定的存储方法,每种产品应有最大允许时间。
灌装前无菌产品的保持时间。
无菌处理时间。
灌装时间。
灭菌容器和封闭容器在关键加工区域(包括灌装)密封前的最大暴露时间。
解读:
EU的GMP对生产过程中的产品保持时间明确的进行了分阶段系统性的考察,从而保证产品的质量。
(2)PDA Process Simulation Testing for Aseptically Filled Products echnical Report No. 22 (2011 Revision) of PDA.
PDA于2011年发布的《Process Simulation Testing for Aseptically Filled Products echnical Report No. 22》关于无菌灌装产品的工艺模拟实验的考量,如下:
2.2Worst Case最差条件
在制药工艺验证中最常采用的技术之一是采用“最差条件”。采用“最差条件”是有意对工艺、系统、设备在更高的挑战条件下进行验证。如果在“最差条件”的挑战下,仍能够达到预期的可接受标准,那么在正常条件下,对系统的可靠性将有更高的信心。工艺模拟实验很容易进行“最差条件”的挑战。下面提供了类型的挑战:
使用的原料、组件和密封在无菌工艺区域保留时间超出范围;
解读:
PDA对无菌灌装产品的工艺模拟实验有着详尽的规定,工艺模拟实验证明了无菌生产工艺生产无菌产品的能力,可以根据此指导原则的出发点并结合产品在实际生产过程中物理及化学变化的考量,从而对药液存放时限进行思考。
2、国内相关指导原则
(1)CDE:《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》
国家药品监督管理局药品审评中心(CDE)于2020-12-31发布的《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》中对药液存放时限有明确规定。如下:
研究确定药液配制后至过滤前的存放时间、药液过滤操作的时间、过滤后至灌装前放置的时间等工艺时限,降低除菌过滤的风险。
解读:
CDE参考了EU、FDA、PDA等相关指导原则并结合实际生产,关注到了无菌工艺生产过程中关键工序的风险点(工序对产品物理、化学、生物学及微生物学的影响),从而对药液存放时限的考察制定了科学合理的规定。
二、药液存放时限的系统性考量
药液存放时限的系统性考量需要依据车间实际生产步骤(生产系统、设备参数及生产组件)指导小试开发,再根据小试开发结果进行放大生产验证,从而形成可持续生产出符合质量要求的产品工艺。
1、药液存放时限的开发
(1)评估生产工序对药液存放时限的风险;
(2)根据可能影响药液存放时限的工艺参数,进行小试开发;
(3)根据小试开发结果进行放大生产验证;
(4)根据放大生产验证的结果指导工艺验证;
(5)形成科学合理的药液存放时限;
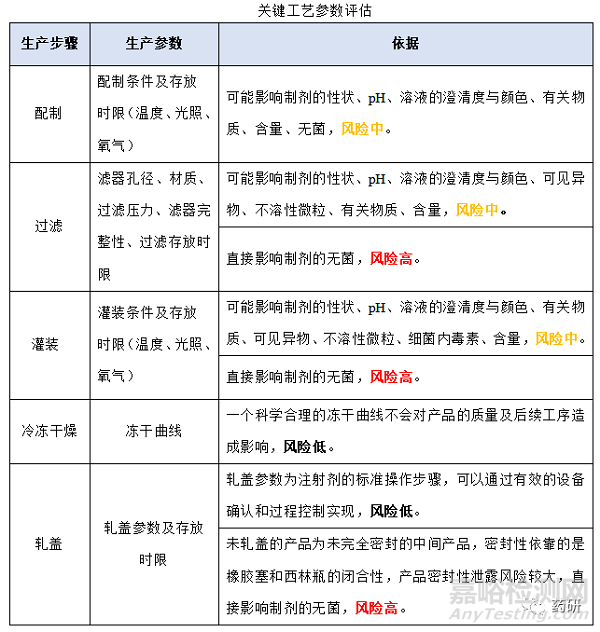
2、生产工序对药液存放时限的风险评估
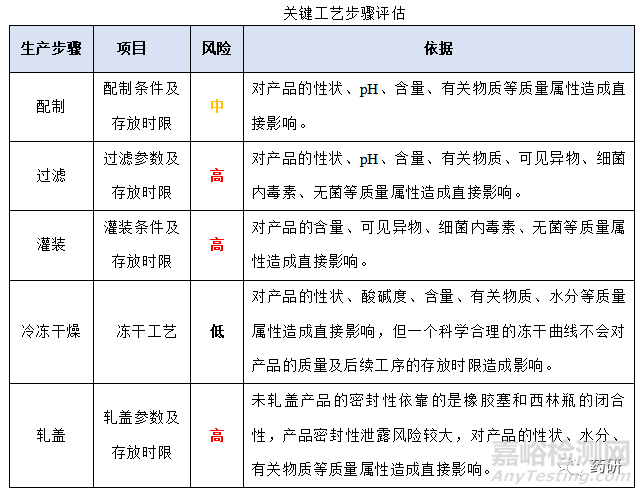
(1)关键工艺步骤评估
(2)关键工艺参数评估
(3)生产过程中的存放时限
(4)确定生产过程中的存放时限
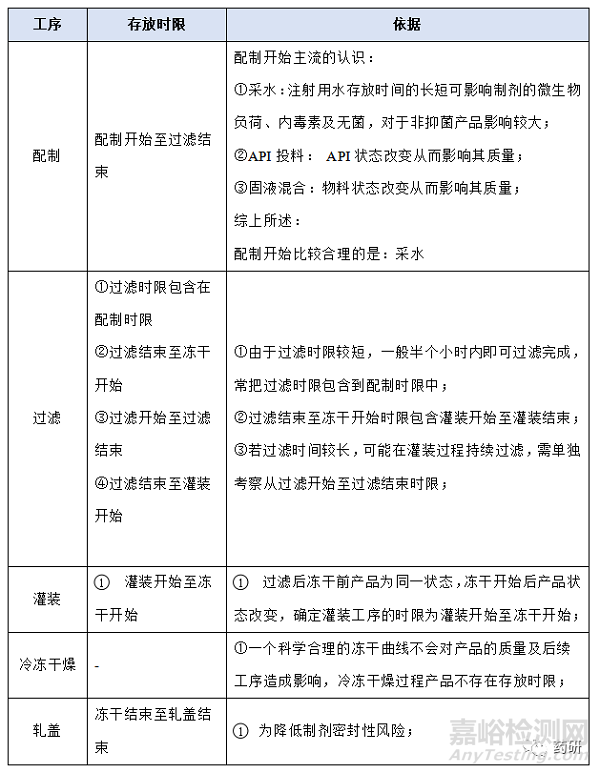
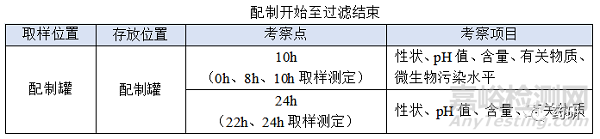
① 配制开始至过滤结束;
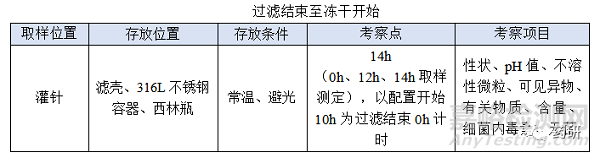
② 过滤结束至冻干开始;
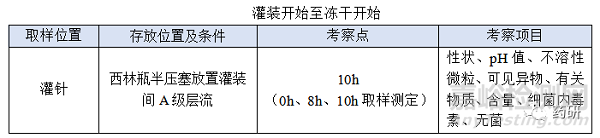
③ 灌装开始至冻干开始;
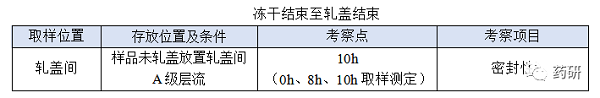
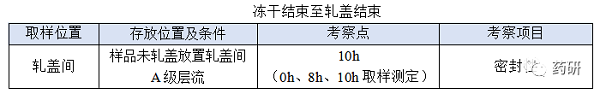
④ 冻干结束至轧盖结束;
3、药液存放时限的小试开发
3.1 配制、过滤及灌装
注意事项:
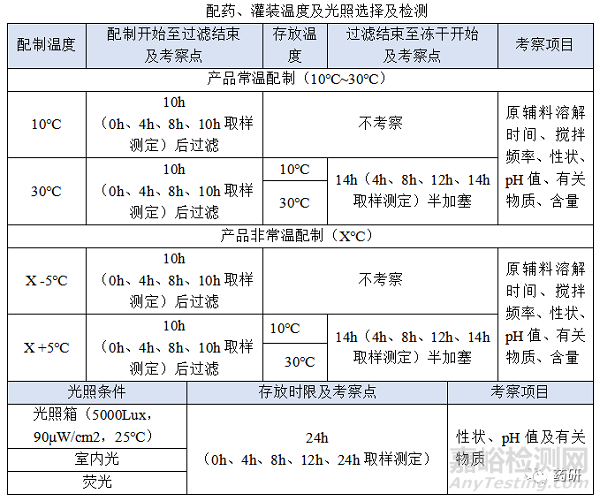
(1)配制开始至过滤结束,温度:常温配制的产品一般考察10℃~30℃,高温X℃配制的产品一般考察X-5℃~X+5℃,特殊品种另外考虑;
(2)配制开始至过滤结束,时间:对于普通冻干粉针剂从配制开始到配置结束一般不超过4h,中间产品检测一般不超过2h,过滤一般不超过0.5h,考察10h是为了应对生产异常,特殊品种另外考虑;
(3)过滤结束至冻干开始,温度:无论是常温配制还是高温配制一般都在配制结束降至常温过滤灌装,低温配制灌装产品、降温后改变药液状态(如黏度)产品等特殊品种另外考虑;
(4)过滤结束至冻干开始,时间:对于普通冻干粉针剂从灌装开始至冻干开始一般不超过8h,过滤结束至冻干开始考察14h是为了应对生产异常,特殊品种另外考虑;
(5)通过对生产过程分析,产品与光照接触的位置有:
① 投料时短暂接触;
② 灌装时短暂接触;
③ 灌装机停机时接触;
④ 灌装完成转移至冻干机短暂接触;
⑤ 冻干机等待开始冻干时接触;
制药生产车间一般采用荧光灯(照度为300lx),小试考察光照箱(5000Lux,90μW/cm2,25℃)、室内光、荧光可覆盖正常生产时的光源,考察时间24h可覆盖产品与光照最长接触时间,特殊品种另外考虑;
3.2 冷冻干燥
不考察
3.3 轧盖
注意事项:
(1)冻干结束至轧盖结束,时间:对于普通冻干粉针剂从冻干结束至轧盖结束一般不超过8h,考察10h是为了应对生产异常,特殊品种另外考虑;
4、放大生产过程中考察药液存放时限
依据小试开发结果设计放大生产中关于药液存放时限的考察,若小试开发上述考察结果皆不影响产品质量,可依据小试开发结果设计放大生产,若小试开发上述考察结果影响产品质量,则重新设计小试工艺。
4.1 模拟存放
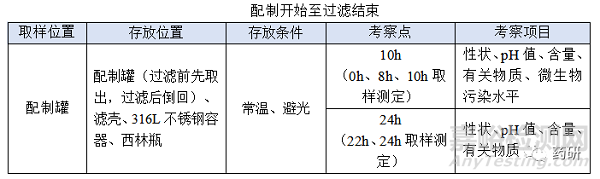
4.1.1 配制
注意事项:
(1)存放容器尽量选择和配制罐材质一致,顶空气体与配制罐一致;
(2)存放条件(温度、光照)与在配制罐一致;
(3)配制开始至过滤结束时限目标定为8h,考察10h留有冗余;
(4)使用经验证的滤器不会改变产品的理化性质,考察22h包含过滤结束至冻干开始12h,考察24h留有冗余;
4.1.2 过滤
注意事项:
(1)时间点的选择需以配置开始10h为过滤结束0h计时;
(2)过滤结束至冻干开始目标定为12h,考察14h留有冗余;
4.1.3 灌装
注意事项:
(1)过滤结束至冻干开始时限包含灌装开始至冻干开始时限;
(2)存放条件与正常生产一致(A级层流);
(3)根据灌装开始的含量确定排液体积;
(4)灌装开始至冻干开始时限目标定为8h,考察10h留有冗余;
4.1.4 冷冻干燥
不考虑时限;
4.1.5 轧盖
注意事项:
(1)存放条件与正常生产一致(A级层流);
(2)冻干结束至轧盖结束时限目标定为8h,考察10h留有冗余;
4.2 真实存放
4.2.1 配制
注意事项:
(1)配制开始后10h过滤;
(2)过滤前将用于22h、24h取样检测的药液取出,待过滤完成倒回配制罐;
4.2.2 过滤
注意事项:
(1)过滤结束14h再开始冻干;
4.2.3 灌装
注意事项:
(1)灌装开始10h再开始冻干;
(2)在冻干结束后取灌装在8h~10h之间的样品用于影响因素或稳定性考察;
4.2.4 冷冻干燥
不考虑时限;
4.2.5 轧盖
注意事项:
(1)轧盖工序时限模拟存放与真实持续相应时间存放无明显差别,可模拟存放;
5 工艺验证过程中考察药液存放时限
依据放大生产过程中药液存放时限考察的结果设计工艺验证药液存放时限,若放大生产上述考察结果皆不影响产品质量,可依据放大生产结果设计工艺验证存放时限,若放大生产上述考察结果影响产品质量,则重新设计放大生产工艺或依据相应数据设计工艺验证药液存放时限。
三、参考资料
1、化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)
2、《中国药典》( 2020年版)中国医药科技出版社, 2020.
3、国家药品监督管理局.关于发布除菌过滤技术及应用指南等3个指南的通告(2018年第85号)
4、EU.GMP Annex 1 : Manufacture of Sterile Products(2020).
5、EMA. Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container(2019).
6、PDA Process Simulation Testing for Aseptically Filled Products echnical Report No. 22 (2011 Revision) of PDA.