摘 要
长效注射剂已逐渐用于抗精神分裂症治疗,棕榈酸帕利哌酮长效注射剂有效控制药物释放,有良好的安全性、临床治疗效果和明显的临床优势,可以一定程度上提高患者依从性。本文通过检索相关国外审评报告和文献资料,概述国内外上市申报的棕榈酸帕利哌酮长效注射剂情况,对此类药物的药学研究进行探讨,提出关注处方工艺和质量控制等研究的考虑,为其研究开发提供一定的参考。
棕榈酸帕利哌酮是帕利哌酮的棕榈酸酯,在体内水解生成帕利哌酮。帕利哌酮为第2代抗精神病药物,是利培酮的活性代谢物9-羟基利培酮,为选择性单胺能拮抗药,对多巴胺D2受体和5-羟色胺5-HT2A受体具有拮抗作用。精神分裂症治疗过程相对较长,部分患者用药依从性差,可能存在自行吐药或者抗拒吞咽情况,一些特殊注射剂可以达到长效或减毒的目的等。作为利培酮代谢产物前药制剂,棕榈酸帕利哌酮长效注射剂利用纳米晶体技术制备,通过剂型的改良满足了临床需求,利于维持长期治疗效果、降低复发率等,具有明显的临床应用价值,也延长了产品的生命周期。
长效注射剂(LAI)可以经皮下或肌内注射等途径给药,在注射位点形成药物储库发挥长效释放作用。LAI能提高药物的便利性(减少给药频率)和治疗效果(控制药物释放和降低药物不良反应发生率)。但同时技术壁垒较高,在药学研究时对辅料、工艺技术和商业化生产能力等具有较高要求,且突破原研专利较难,较难被仿制。
本文结合上市药物的相关专利和文献,针对棕榈酸帕利哌酮长效注射剂从处方工艺研究、质量研究控制等方面探讨本品仿制药药学研究可以关注的问题。
1、棕榈酸帕利哌酮长效注射剂的国内外上市情况
1.1 美国上市情况
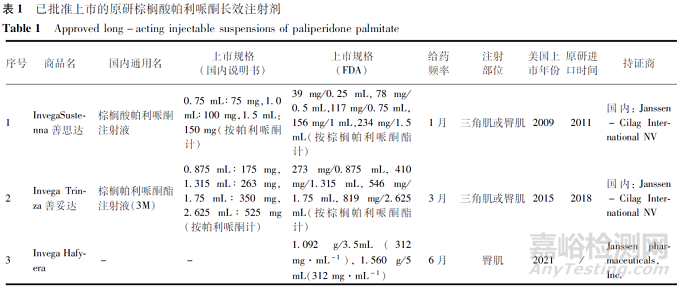
棕榈酸帕利哌酮由杨森公司开发,目前上市原研制剂包含1月制剂(PP1M)、3月制剂(PP3M)和6月制剂(PP6M)。PP1M(商品名Invega Sustenna,NDA022264)于2009年在美国上市,上市5个规格。PP3M(商品名Invega Trinza,NDA0207946)于2015年在美国上市,上市4个规格。PP6M(商品名Invega Hafyera)于2021年在美国上市,上市2个规格(见表1)。
美国食品药品监督管理局(FDA)鼓励仿制长效注射剂的研发,可以在一定程度上使患者更好地获得可负担的药物。FDA发布了PP1M和PP3M的个药指南(PSGs)以指导仿制药研发,2022年FDA计划修订的个药指南列表中包含PP1M,将对体内研究设计做微小的修改。
2021年7月FDA批准了以Invega Sustenna(NDA022264)为参比制剂(RLD)的首个仿制棕榈酸帕利哌酮注射液,仿制药(ANDA)编号为211149,持证商为Teva公司,暂未销售。FDA在2021年度仿制药报告中提到此仿制药使用了FDA开发的用于药代动力学研究设计和生物等效性评估的模型和模拟方法。其FDA批准信中说明原研存在专利保护期(美国专利的最后到期日2031年),仿制企业在ANDA申报资料中包含了专利第Ⅳ段声明,截至2022年11月暂未获得180天仿制药市场独占期。FDA公布的第Ⅳ段声明列表(更新日期2022-10-03)中也显示目前有1家仿制药企业针对Invega Sustenna提交了此声明,“180天状态”为“推迟”。
1.2 欧盟和日本上市情况
欧盟已上市原研PP1M(商品名Xeplion),规格为25、50、75、100和150 mg(按帕利哌酮计),和上市原研PP3M(商品名Trevicta),规格为175、263、350、525 mg(按帕利哌酮计)。欧洲已有多家PP1M仿制药通过互认程序(MRP)批准上市,各国间产品名存在差异,例如Dopelir参考成员国(RMS)为丹麦,Paliperidone Biogaran的RMS为法国。日本也上市了原研PP1M(Xeplion)和PP3M(Xeplion TRI)。
1.3 中国上市申报情况
国内仅有原研产品PP1M和PP3M上市,分别上市了3个规格和4个规格(见表1)。截至2022年12月底,暂无国产制剂上市,国内已有2家国产企业申报PP1M上市、1家国产企业申报PP3M。
国家药品监督管理局药品审评中心发布的《化学仿制药参比制剂目录》(截至2022年11月第57批)已公布棕榈酸帕利哌酮长效注射剂的多个参比制剂,包括原研进口和未进口原研药品(美国橙皮书、欧盟上市),包含PP1M 13个、PP3M 12个,目前暂未有PP6M的参比制剂公布。仿制药企业首先可以结合品种情况选择合适的参比制剂。
2、药学研究探讨
2.1 处方工艺
国外的[FDA、欧洲药品管理局(EMA)和日本药品和医疗器械管理局(PMDA)]审评报告和说明书显示PP1M处方组成包括无菌原料药、聚山梨酯20、聚乙二醇4000、一水柠檬酸、无水磷酸氢二钠、磷酸二氢钠一水合物、氢氧化钠和注射用水,PP3M和PP6M缓冲盐中去除了无水磷酸氢二钠。基于药代动力学模拟,PP3M剂量是PP1M的3.5倍,同时为了减少给药体积,PP3M中原料药浓度由PP1M的156 mg·mL-1(以棕榈帕利哌酮酯计)增加到321 mg·mL-1,辅料的用量有一定的差别。FDA说明书中PP6M还明确了各辅料的用量,聚山梨酯20(10 mg·mL-1)、聚乙二醇4000(75 mg·mL-1)、一水柠檬酸(7.5 mg·mL-1)、磷酸二氢钠一水合物(6 mg·mL-1)、氢氧化钠(5.4 mg·mL-1)和注射用水,较PP3M增加了注射体积。FDA和EMA的PP3M审评报告中说明了不同规格存在过量灌装情况。对于特殊注射剂,仿制药处方原则上应与参比制剂一致。若存在过量灌装需要通过与参比制剂进行对比等研究提供充足的依据,比如可以进行仿制药与参比制剂可抽取药量的对比等。
关于原料药的性质对制剂的影响,FDA和EMA的审评报告显示,棕榈酸帕利哌酮以晶型A稳定存在,在较宽的pH内和水中几乎不溶,这种低溶解度原料药使制备长效混悬肌内注射剂成为可能。此长效注射剂的释药机制主要为在进行肌内注射后,棕榈酸帕利哌酮在注射部位缓慢溶解释放,代谢为帕利哌酮,进一步被循环系统吸收从而发挥作用。影响制剂体内生物等效的关键是注射部位棕榈酸帕利哌酮的溶解行为。
根据本品的释药机制可知,原料药的粒度和粒度分布是影响产品体内释放和起效的关键因素。EMA报告显示PP1M生产过程中采用无菌粉碎来减小原料药粒径,以达到合适的粒径范围,而PP3M通过控制工艺增大原料药粒度,使药物体内溶解和释放速度变慢。PP3M由于与PP1M的给药量、粒径、给药体积的不同,有更慢的释药速度。粒径控制方面建立了经过验证的粒径测定方法,能够准确测定生产、释放和贮藏过程中粒径的变化。建立了针对不同粒径有区分性的体外释放的方法,并进行了粒径和释放的体内外相关性研究(IVIVC)。EMA报告显示PP3M提供了新的体内外相关性的研究数据(因为PP3M的粒径不在PP1M 的IVIVC已验证的范围内),其临床研究显示一定范围内混悬剂粒径的增大可以降低Cmax和延长Tmax。对原料药其他的理化性质,如纯度、结晶性和形态学等进行了研究,进一步评价原料药关键质量属性对制剂特性的影响。
在制剂开发中适宜的辅料也起着重要作用,其决定了混悬液是否具有适宜的溶液外观、pH值、黏度、渗透压、再分散性和物理化学稳定性等。原研专利显示聚山梨酯20属于润湿剂,能够降低水和原料的表明张力;聚乙二醇4000作为悬浮剂,在保持一定的相对用量时使原料药悬浮分散在介质中;使用无水磷酸氢二钠和磷酸二氢钠一水合物来做为缓冲剂调节等渗和中性的环境,且使其中的悬浮酯不易于絮凝。EMA报告显示氢氧化钠做pH调节剂。同时由于考虑给药方便性,需混悬液保持合适的黏度,使短时间内在较细的针头中较易通过。EMA的PP3M可重悬浮性研究采用3批已在室温储存了9至11个月的验证批次进行,并且在说明书中详细说明用前振摇的方法。
工艺开发方面,EMA审评报告中显示制备过程主要包括将无菌原料药分散在无菌介质中,混悬液进行湿法研磨达到目标粒径,用注射用水稀释后进行无菌灌装。拟定的关键步骤为除菌过滤、研磨和灌装,同时制定了一系列的过程控制措施。对于无菌生产工艺,仿制药处方工艺开发中应充分考虑各生产步骤的无菌控制。在工艺描述时如果能够充分描述各步骤,特别是除菌过滤、研磨和灌装的工艺参数更有利于理解工艺过程,例如除菌过滤可以明确过滤器材质、型号等信息,并监测其过滤前后完整性和过滤压力等参数,比较容易遗漏除菌过滤前料液的微生物负载情况。研磨步骤可以关注研磨机转速、研磨时间、研磨介质使用情况(如介质装量)等参数。进行无菌工艺验证注意可以包含培养基灌装试验,直接接触无菌物料和产品的设备、部件、容器密封性系统的除菌除热原验证等。目前湿法研磨技术较为成熟并得到了广泛使用,市场上也有不同品牌的纳米研磨机可供研究和生产使用。关于批量问题,针对此类特殊注射剂注册批和商业批的批量原则上应保持一致。
包材选择方面,原研制剂内包装均采用透明的环烯共聚物(COC)预灌封注射器(PFS)(连接带胶塞推杆、护帽),配有逆止器和安全针头。EMA审评报告中显示申请人已对包材的相容性和密封性均进行了详细研究。使用中稳定性研究则对给药过程中的功能性进行了研究,包括针头与注射器对拆离力矩(PRF)、轴向分离力(PTF)。仿制药是否选择与参比制剂相同的包材,需对选择的包材提供充分的依据,也可以关注粒度与针头的匹配性。
2.2 质量研究与质量标准
长效注射剂除需要满足常规注射剂的基本要求外,通常会结合制剂特点和自身释药特性等有一些特殊研究内容。EMA的PP3M审评报告中在质量控制方面对性状/可重悬浮性/可注射性、鉴别(HPLC、IR)、pH(6.5~7.5)、有关物质、含量均匀度、不溶性微粒、体外溶出、粒度分布、无菌、细菌内毒素和含量进行了研究。
药品审评中心发布的《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》中针对特殊注射剂的质量属性方面有一些考虑。考察的关键质量属性可能包括但不限于以下内容:理化性质(如性状、黏度、渗透压摩尔浓度、pH值/酸碱度等),Zeta电位,粒子形态,粒径及分布,体外溶出/释放行为,药物晶型和结晶形态。仿制药可以参考此技术要求结合品种特性对部分质量属性进行研究,原则上应提供至少3个批次参比制剂样品的质量对比考察数据。例如针对此长效制剂,黏度不但会影响稳定性(过低的黏度会加速药物晶体的沉淀),对通针性和给药器内药物残留等均有影响。
溶出度作为关键质量控制指标,国外审评报告显示原研制剂进行了IVIVC研究。目前未查询到此类长效注射剂在药典中或者通用的体外溶出方法,而且建立IVIVC也面临一些挑战。仿制制剂可以在生物等效性试验前在体外考察自制样品与参比制剂溶出相似情况,一定程度上降低生物等效性不等效风险(但有时体内外相关性并不好);标准中增加此控制可以评价自制商业批样品与关键批次(例如临床试验批次)的质量批间一致性等。通常采用多点法制定研究限度,其各阶段时间点选取和限度的设置可以考虑结合仿制制剂自身特性和多批关键批次(例如临床批次)的质量研究和稳定性数据等拟定,提供充分合理的依据,而非仅参考原研制剂拟定的标准。FDA溶出度数据库中提供了可供参考的PP1M和PP3M的溶出方法:桨法,转速50 r·min-1,溶出介质为含0.489%吐温20的0.001 mol·L-1的HCl,溶出介质体积为900 mL,PP1M的取样时间为1.5、5、8、10、15、20、30和45 min,PP3M的取样时间为5、30、60、90、120、180、240、300和360 min。
2.3 稳定性研究
EMA的PP3M审评报告中指出稳定性研究的考察指标包括性状/可重悬浮性/可注射性、粒度分布、体外溶出、pH、有关物质、无菌、细菌内毒素和含量,另外增加了醛含量、质量损失和容器密封性指标。采用正置和倒置不同的放置方式,稳定性考察条件包括了长期条件(25 ℃/40% RH、30 ℃/35% RH)、中间条件(30 ℃/75%RH)和加速条件(40 ℃/不超过25%RH)。在50 ℃强力试验条件下时观察到粒径的明显上升且体外释放降低,这是由于奥斯特瓦尔德熟化现象。这种粒径增长现象也可以在加速6个月(40 ℃/25%RH)和长期12个月(30 ℃)的情况下观察到。在冻融或者冷冻情况下,可以观察到粒径在初期明显下降。原研制剂说明书中明确了储存温度且说明请勿冷冻保存。PP3M最小灌装量(0.875 mL)样品在6月(40 ℃/25%RH)有轻微失水情况,可能是由于半透的注射器造成。仿制药在进行稳定性研究时可以结合自身包材选择情况等按照ICH指导原则拟定合理的考察条件。
混悬型注射剂会发生奥斯特瓦尔德熟化现象,即小粒子溶解并分布在大粒子表面,小粒子逐渐减少和粒度的增长,导致混悬剂比表面积减少,并进一步导致溶出度的下降(药物的溶解度与颗粒粒度大小呈反比)。而高温一般增加了药物晶体在悬浮液中的溶解度,更易加剧此现象的产生。混悬型注射剂开发时通常会从优化制剂组成(如降低共溶剂或表面活性剂的水平、增加黏度、缩窄粒度分等)和控制产品的储存条件等方面,减少奥斯特瓦尔德熟化现象的发生。仿制药处方工艺筛选过程也可以进行适当的稳定性考察,进一步确认原料药粒径分布选择范围、辅料型号等选择的合理性。
3、结 语
此纳米晶混悬注射液对难溶性药物有高药物负荷的优点,且可以使用小针头注射,使药物局部集中,可最小化组织损伤的风险;其可以减少复发和用于治疗的维持阶段等进而提高疗效和治疗依从性,因为诸多优势,抗精神病领域基于纳米晶体技术的长效注射液获得较大市场和巨大的商业价值。但是工业界也面临诸多挑战,如生产问题(扩大生产、无菌保障水平、可注射性等)、IVIVC建立(长效注射剂比口服制剂有更差的体内外相关性,获得A级相关更为困难)等。目前国内暂无国产榈酸帕利哌酮长效注射剂获准上市,其具有良好产业化前景,值得业界开发应用和探讨,以更好地造福患者。