您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-10-13 06:47
甲苯咪唑是一种广谱的驱虫药,对蛔虫、蛲虫、钩虫和鞭虫感染的治愈率很高。甲苯咪唑因其活性广泛,高效及方便服用而广泛应用,被WHO作为抗肠内蠕虫感染列入基本药物目录。
该药物不溶于水,其同质多晶的研究显示3种晶型(A、B、C)溶解性和治疗的差异。溶解度试验结果显示,在未加表面活性剂的情况下,溶解度由小到大依次为A、B、C晶型。
晶型C因其充分溶解保证最佳生物利用度且没有与晶型B相关的毒性表现而成为制药首选,且一系列的药理研究表明,若要达到预期的药理活性,晶型A的含量不能超过30%,为达到更好的临床疗效,应控制并监测市场上疗效各异的甲苯咪唑片中的晶型形态。
国内对化学药的晶型影响制剂的研发及稳定性的研究日益增多。红外光谱法是以红外区域电磁波连续光谱为辐射源照射样品,记录样品吸收曲线的一种光学法,是有机化合物分析中广泛应用的方法。红外光谱法对被测样品纯度要求较高,一般不低于90%~95%。
在各国药典中,大多数药物原料药的鉴别均采用了红外光谱法,制剂由于辅料的干扰,红外鉴别应用较少。但是USP和BP中不仅原料药广泛采用红外光谱法鉴别,制剂也较多采用红外光谱法进行鉴别。
《中华人民共和国药典》2010年版二部将红外光谱法列为制剂鉴别项的品种仅为73个,约占红外光谱法鉴别总数的11%。
而英美药典制剂采用红外光谱法鉴别的品种数在用该法鉴别总数中在2009年之前所占百分比已高达30%左右。药物制剂采用红外光谱鉴别有专属性强的优点,应更多地开发红外光谱在药物制剂鉴别中的应用,加快我国药品标准国际化进程,提高药品标准。
因制剂过程中加入的各种辅料对制剂中活性药用成分晶型的测定存在不同程度的干扰。
差示扫描量热法(differentialscanningcalorimetry,DSC)不能用于区分制剂中活性药用成分晶型,因为辅料存在热学事件重叠的干扰,导致曲线复杂难以分析;
X射线粉末衍射(X-raypowderdiffraction,XRPD)因辅料的干扰,无法完全识别各晶型的特征峰;
傅立叶变换红外光谱(Fouriertransforminfraredspectrum,FTIR)直接压片法亦受辅料特别是蔗糖、糖精钠的影响,红外光谱难以辨析;
曾尝试使用多种有机溶剂提取制剂甲苯咪唑,测定其红外光谱均为混合晶型,主要原因在于甲苯咪唑溶解后破坏了原来的晶格结构,故使用有机溶剂提取甲苯咪唑并测定红外光谱的方法亦无法真实反映制剂中活性药用成分的晶型形态。
本文通过摸索和分析,利用原辅料溶解度的差异,克服辅料干扰及抑制晶型转化,首次建立了科学、简单的前处理方法——分次水洗并减压干燥,采用红外分光光度法,鉴别甲苯咪唑片/咀嚼片中活性药用成分的晶型,并结合溶出度测定法对国内外的甲苯咪唑片进行验证。
建立简单易行的方法监测国内外市场上疗效各异的甲苯咪唑片/咀嚼片所投原料的晶型形态,弥补现行检验的不足。
1 试药与仪器
1.1 试药
甲苯咪唑原料及片剂/咀嚼片(规格100mg·片 -1)来自WHO和2016年国家抽验评价收集到的样品,其中原料A晶型与C晶型均来自国内的1家原料生产公司,甲苯咪唑片/咀嚼片来自6个制剂厂家,其中国内3家,批号GN001、GN002、GN003,国外3家,批号GW001、GW002、GW003。
1.2 仪器
ThermoNicoletAVatar360红外分光光度计,溴化钾压片法测定,扫描范围为4000~400cm -1。Waterse2695高效液相色谱系统。天津天大天发RC8MD溶出试验仪。
2 方法与结果
2.1 红外光谱
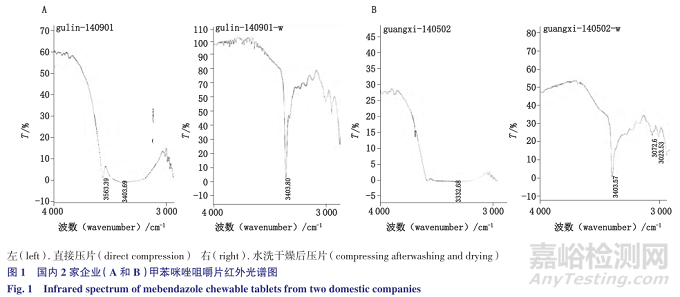
根据文献尝试直接压片,发现国外的样品辅料干扰不明显,国内的2家企业存在严重的辅料干扰,见图1;
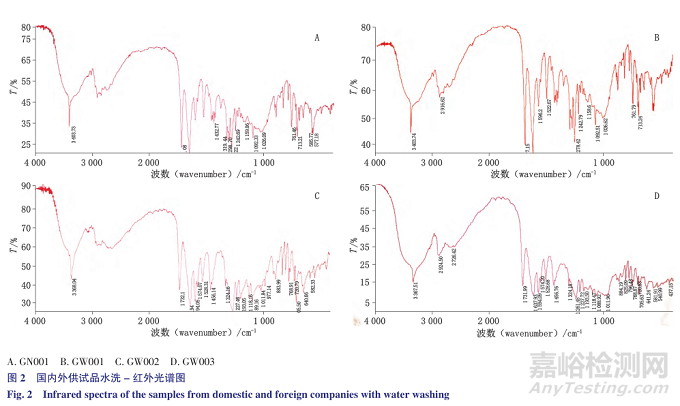
利用甲苯咪唑不溶于水,与极性辅料的溶解度差异,将供试品研细后分2次各加水50mL搅拌均匀,滤过,取滤渣25℃减压干燥1夜,溴化钾压片,进行红外扫描,基本可消除水溶性辅料(蔗糖,糖精钠、色素等)的干扰,在甲苯咪唑3种晶型的特征吸收所在4000~1000cm -1波数范围内,与标准图谱差别不大,能够明确区分甲苯咪唑的晶型,反映出企业采用的原料晶型及制剂过程中是否发生转晶的现象。
结果国内3家生产企业均为晶型C投料,而国外有2家为A型投料,1家为晶型C投料。典型红外图谱见图2。甲苯咪唑3种晶型的典型红外吸收峰(-NH及-C=O)波数列表见表1。

2.2 结果验证
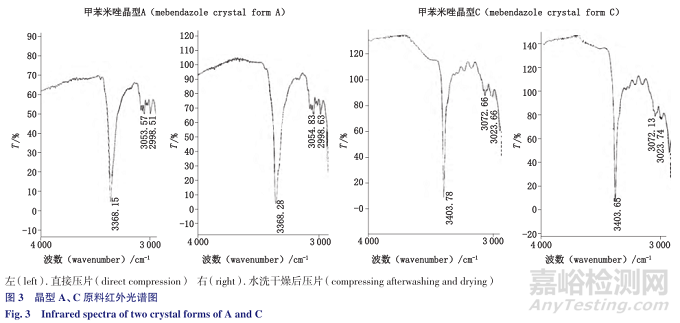
有文献证实甲苯咪唑在高温高湿条件下会发生晶型转变的现象,但条件相当苛刻,将甲苯咪唑的晶型C与水制成混悬液,在100℃下放置5h,转成晶型A的比例未超过1%。
通过试验验证,甲苯咪唑晶型A、C原料同法处理之后,溴化钾压片,与直接压片的结果无明显差异,见图3。
说明水洗减压干燥的前处理过程未发生转晶。本法既可去除部分辅料干扰,又能保证晶型不转化,达到简单有效鉴别制剂中活性药用成分晶型的目的。
2.3 溶出度验证
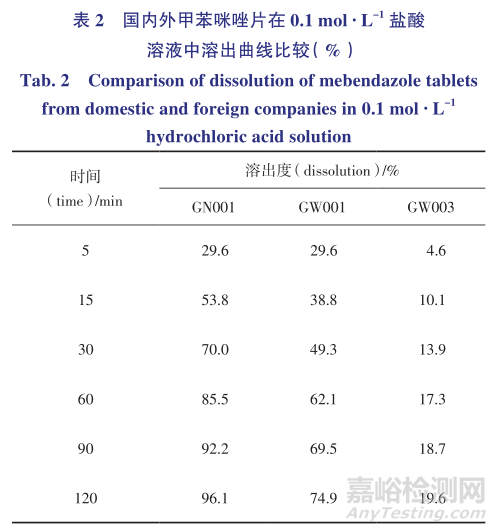
文献报道甲苯咪唑3种晶型在0.1mol· L-1的盐酸溶液中的溶出量分别为晶型A20%(120min)、晶型B37%(120min)、晶型C70%(120min),因此在美国药典溶出度试验的基础上去除了其溶出介质中的十二烷基硫酸钠,仅用0.1mol· L-1的盐酸溶液作为溶出介质以区分甲苯咪唑片中的活性药用成分晶型。
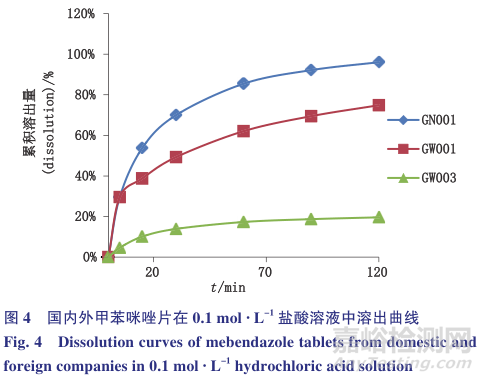
因《中华人民共和国药典》2015年版对咀嚼片没有进行溶出度试验的要求,选取国内外共3家企业规格均为100mg的甲苯咪唑片进行试验,比较其在0.1mol· L-1的盐酸溶液中的溶出曲线,结果见表2和图4。
国内的甲苯咪唑片能基本溶出(120min),国外1家企业的甲苯咪唑片溶出75%(120min),而另外一家的甲苯咪唑片溶出量少于25%(120min),与3家企业的红外晶型鉴别的结果相吻合,说明0.1mol· L-1盐酸溶液对甲苯咪唑片溶出度有一定区分力,可以反映制剂在处方工艺和晶型方面的差异。
3 讨论与结论
本文通过摸索分析,克服辅料干扰,抑制晶型转化,建立了科学简便的前处理方法,采用快捷,具选择性的红外分光光度法鉴别甲苯咪唑制剂中活性药用成分的晶型,顺应国际标准的导向趋势,亦可满足快速检验的需求,已为2017年版《国际药典》所收载;
并验证了未添加表面活性剂的溶出介质用于区分甲苯咪唑片中活性药用成分晶型的差异性,二者结合,相辅相成,为监控国内外市场上疗效各异的甲苯咪唑片/咀嚼片中活性药用成分的晶型提供简便易行的方法。

来源:《药物分析杂志》


