您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-08-21 18:56
摘要
靶向蛋白降解药物通过特异性降解目标蛋白, 为不同疾病的治疗提供了新的策略。然而, 基于小分子的传统降解剂仍然面临生物利用度差、E3泛素连接酶可用性受限及脱靶效应等显著挑战。基于生物大分子的靶向蛋白降解药物拥有更灵活的设计和更高的特异性, 有望克服小分子蛋白降解嵌合体药物的局限性。本文聚焦生物大分子降解体系, 基于细胞内两大蛋白的核心降解通路(泛素−蛋白酶体途径与溶酶体途径), 系统地分析比较不同技术路线的分子作用机制、临床转化优势以及潜在挑战, 并探讨跨膜蛋白降解、光控蛋白降解等前沿进展及未来发展方向, 为构建基于精准靶向蛋白降解的治疗体系提供思路。
关键词
靶向蛋白降解; 生物大分子降解体系; 生物蛋白酶靶向嵌合体; TRIM-Away
近年来, 蛋白水解靶向嵌合体(proteolysis-targeting chimeras, PROTACs)、分子胶等靶向蛋白降解药物研发技术发展迅速, 致力于解决传统抑制剂药物的缺陷。PROTACs和分子胶通过连接靶蛋白与E3泛素连接酶触发泛素−蛋白酶体降解系统。与主要通过占位驱动的传统小分子相比, PROTACs通过“事件驱动”模式实现靶蛋白的完全降解, 无需持续占据活性位点便能直接清除无明确结合口袋的蛋白。然而, 这类基于小分子的靶蛋白降解药物仍面临许多挑战, 比如可利用的E3泛素连接酶种类有限, 以及脱靶毒性高等问题。
为突破上述局限, 研究人员把目光转向基于生物大分子的靶蛋白降解药物。基于生物大分子的靶蛋白降解药物将生物大分子(如抗体、多肽、核酸等)作为核心功能单元, 通过特异性识别致病蛋白, 利用生物大分子和靶蛋白的高亲和力, 实现对致病蛋白的精准清除。相较于小分子药物, 生物大分子降解剂在靶向精度和多功能性方面展现显著优势(表1)。本文综述了近年来具有代表性的基于生物大分子的靶蛋白降解策略, 深入讨论其优势与挑战, 并对未来的临床应用进行展望。
1靶蛋白降解途径
靶向蛋白降解技术主要利用细胞内两大蛋白降解途径: 泛素−蛋白酶体系统(ubiquitin-proteasome system, UPS)途径与溶酶体途径。从功能特性分析, 这两个系统在降解机制(泛素依赖与非泛素依赖)和生理适用性(胞质可溶性蛋白与膜结合或聚集态蛋白)等层面具有显著区别。
1.1 泛素−蛋白酶体系统途径
Hershko等首先基于无细胞体系与酶分离系统的机制研究获得关键突破, 后续该实验室基于哺乳动物细胞及酵母模型的研究与其形成机制互补, 并证实活细胞内绝大多数蛋白质降解过程依赖于泛素分子的共价偶联机制。接着, Varshavsky进一步阐明了泛素化级联反应的分子机制: E1泛素激活酶首先通过硫酯键(Cys-UB)激活泛素, 消耗ATP生成AMP和PPi。接着, 活化的泛素从E1泛素激活酶转移到E2结合酶活性中心的半胱氨酸残基。最后, E3泛素连接酶催化泛素从E2转移到靶蛋白的特定赖氨酸残基, 形成以K48连接为主的多聚泛素链。泛素化靶蛋白可以被蛋白酶体识别, 蛋白酶体将其去折叠并降解为短肽。
1.2 溶酶体途径
溶酶体途径介导的靶蛋白降解途径主要通过两条核心途径实现: 内体−溶酶体途径和自噬−溶酶体途径。①内体−溶酶体途径: 负责降解通过内吞作用摄入的细胞外物质(如配体−受体复合物、病原体)以及膜蛋白的循环与降解; ②自噬−溶酶体途径: 通过自噬清除细胞内受损或冗余成分(如线粒体、蛋白聚集体), 在应激(如饥饿、氧化压力)时提供能量和原料。
2基于生物大分子的靶向蛋白降解药物
2.1 蛋白酶体介导的降解途径
2.1.1 生物蛋白酶靶向嵌合体
生物蛋白酶靶向嵌合体(biological proteolysis-targeting chimeras, bioPROTACs)技术是在PROTACs技术框架上的创新延伸。传统PROTACs通过化学连接子将靶蛋白配体与E3泛素连接酶招募配体相连, 形成异源双功能分子, 诱导靶蛋白与E3连接酶接近, 进而促使靶蛋白泛素化并被蛋白酶体降解。然而, 传统PROTACs在应用中受到E3连接酶可用性有限以及脱靶毒性问题的制约, 仅少数E3连接酶(如Cereblon和Von Hippel-Lindau protein)被成功应用于PROTACs设计, 极大地限制了可靶向蛋白质的种类, 且非特异性结合导致的脱靶降解可能干扰正常细胞功能, 引发毒性反应。
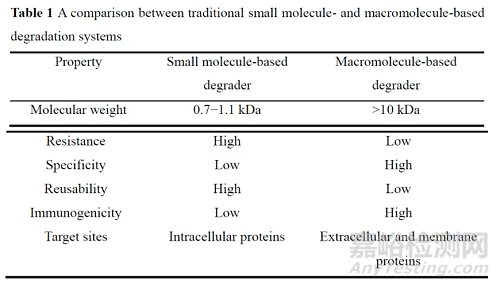
为突破传统PROTACs的技术限制, Lim等系统探索了bioPROTACs。该技术基于工程化重组策略, 将E3泛素连接酶招募域与靶蛋白结合域直接共价融合, 构建无连接子结构的单链蛋白复合体。如图1所示, 其作用机制为: 当bioPROTACs同时结合靶蛋白和E3泛素连接酶时, E3泛素连接酶促使泛素分子通过级联反应共价修饰靶蛋白。泛素化标记的靶蛋白随后被转运至蛋白酶体进行特异性降解。
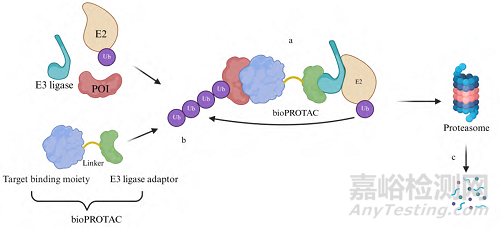
Figure 1 The mechanism of bioPROTACs. a: Recognition and binding, the bioPROTAC simultaneously binds to the target protein and E3 ligase; b: Ubiquitination of target proteins, E3 ligase prompts the ubiquitin molecule to modify the target protein; c: Proteasome recognition and degradation, the ubiquitin-labeled target protein is then transported to the proteasome for specific degradation. E2: E2 ubiquitin-conjugating enzyme; POI: Protein of interest; bioPROTAC: Biological proteolysis-targeting chimera; Ub: Ubiquitin
bioPROTACs技术在靶蛋白降解的高效性和特异性方面展现出显著优势。Lim等的研究表明, Con1-SPOP bioPROTAC能够高效降解增殖细胞核抗原融合蛋白, 如细胞核抗原−绿色荧光蛋白(proliferating cell nuclear antigen-green fluorescent protein, PCNA-GFP)和内源性增殖细胞核抗原(proliferating cell nuclear antigen, PCNA)。与RNA干扰(RNA interference, RNAi)相比, 其起效时间大幅缩短, 且不依赖于蛋白质的自然周转, 为降解长寿命蛋白(如PCNA)提供新的解决方案。此外, 该团队开发的基于SPOP E3泛素连接酶的anti-RAS bioPROTACs实现了对RAS蛋白的选择性降解, 进一步证实了bioPROTACs的高特异性。
然而, 这类生物大分子在具有以上优势的同时, 仍然在递送效率和分子稳定性方面存在挑战: ①其跨膜运输效率受细胞膜屏障严重制约, 当前研究重点集中于开发生物相容性脂质纳米颗粒等新型递送载体, 以改善其膜渗透性与胞内递送效率; ②部分核酸或蛋白质组分的bioPROTACs较易被血清核酸酶或蛋白酶降解, 可能需频繁高剂量给药, 带来不良反应风险; ③靶蛋白的泛素化位点不易被小分子药物遮蔽, 但可能会被大分子bioPROTACs所遮蔽, 导致不可成药。为解决该问题, 未来可尝试仿照Bridged PROTAC的思路, 使药物间接靶向目标蛋白, 从而绕开可能被遮蔽的泛素化位点, 提高降解效率。
综上所述, bioPROTACs作为一种新兴的靶向蛋白降解技术, 在克服传统PROTACs局限性方面展现出巨大潜力, 但在递送效率、分子稳定性及靶向精准性等方面仍需进一步优化, 以推动其在临床治疗中的广泛应用。
2.1.2 TRIM-Away
TRIM-Away技术作为一种新兴的靶向蛋白降解策略, 凭借其快速起效和高特异性的优势, 在靶向蛋白降解领域受到广泛关注。该技术的核心机制基于细胞内源性TRIM21蛋白的泛素化功能, 通过特异性抗体与靶蛋白结合, 进而利用TRIM21介导的泛素化修饰, 将靶蛋白导向26S蛋白酶体进行降解。这一过程不仅高效且具有高度选择性, 为靶向降解胞内蛋白提供了全新的思路。
TRIM-Away技术的实现依赖于两个关键步骤(图2)。首先, 通过显微注射或电穿孔等技术将特异性抗体导入细胞内, 抗体通过其抗原结合位点与靶蛋白形成高亲和力复合物; 随后, 细胞内的TRIM21蛋白通过其PRYSPRY结构域特异性识别抗体的Fc段, 进而介导抗原−抗体复合物的泛素化修饰, 并最终将复合物导向26S蛋白酶体进行降解(图2A)。
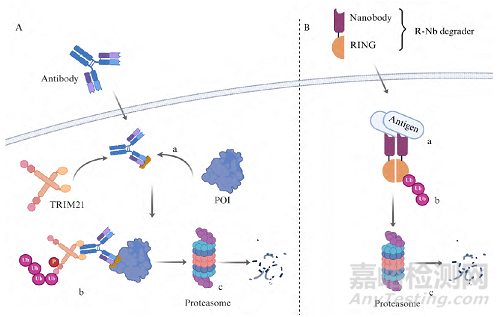
Figure 2 Schematic illustration of TRIM21-based protein degradation mechanism.
A-a: Formation of the complex. Specific antibodies are delivered into cells, and the antibodies form high-affinity complexes with the target proteins; A-b: Ubiquitination of RING structure of the complex. The TRIM21 protein specifically recognizes the Fc segment of the antibody, mediates the binding of the antigen-antibody complex, and catalyzes the ubiquitination modification; A-c: Degradation of the complexes in the proteasome; B-a: Formation of the complex. Specific nanobodies are delivered into cells form high-affinity complexes with the target antigen; B-b:Ubiquitination of RING structure of the complex; B-c: Degradation of the complexes in the proteasome. P: Phosphate group; R-Nb: RING-nanobody
TRIM-Away技术的高特异性源于其双重识别机制。一方面, 抗体与靶蛋白的特异性结合确保了对目标蛋白的精准识别; 另一方面, TRIM21对抗体Fc段的特异性识别进一步提高了系统的整体特异性。这种双重筛选机制显著降低了非特异性结合的可能性, 从而减少了脱靶效应。Clift等在小鼠卵母细胞和培养细胞中制备和递送抗体, 靶向降解运动蛋白-5(kinesin-5, Eg5), 证实了TRIM-Away的高特异性。
近年来, 随着TRIM-Away技术不断发展, Benn等构建了基于TRIM-Away机制的融合蛋白。该研究团队将TRIM21的RING结构域与靶向目标蛋白的纳米抗体融合, 构建融合蛋白, 利用纳米抗体靶向目标蛋白, 通过RING结构域催化泛素从E2酶转移到目标蛋白上, 从而标记目标蛋白并使其降解(图2B)。他们将TRIM21的RING结构域与靶向tau蛋白的纳米抗体融合, 构建了RING纳米抗体(RING-nanobody, R-Nb)降解剂。利用TRIM21的聚集依赖性, R-Nb化合物结构被证明可以降解不溶性tau聚集体, 同时可溶性单体tau基本不受影响。
由此可见, TRIM-Away在神经退行性疾病的治疗领域展现出很大潜力。中枢神经系统内特定蛋白质病理性沉积是阿尔茨海默病、帕金森病及亨廷顿病等神经退行性疾病的重要诱因之一。TRIM-Away特异性、选择性地降解聚集蛋白且不干扰单体蛋白为其提供了有效的治疗方案, 在提高药物特异性和选择性的同时降低了药物毒性。
然而, TRIM-Away技术依然存在诸多挑战, 限制了其临床应用: ①大分子质量传统抗体存在透膜效率低、递送效率低等问题, 导致靶蛋白降解效率受限; ②核定位受限, 所以针对核内蛋白质的降解可能需要更高浓度的抗体, 提高了抗体生产的工艺难度; ③长时间使用较高浓度的抗体可能诱发抗药物抗体(anti drug antibody, ADA)反应。
针对未来的研究方向可能包括: ①开发新型递送系统, 利用生物相容性脂质纳米颗粒、病毒载体等新型递送系统, 提高抗体的细胞内递送效率; ②通过人源化抗体, 通过保留人源框架区(framework region, FR), 以降低ADA反应的发生率; ③通过优化抗体设计, 开发更小分子质量的抗体或抗体片段(如纳米抗体), 以提高其透膜效率和细胞内分布; ④拓展应用范围, 探索TRIM-Away技术在其他疾病领域的应用, 如癌症和代谢性疾病, 进一步验证其临床潜力。
综上所述, TRIM-Away技术作为一种高特异性的靶向蛋白降解策略, 在神经退行性疾病等领域的治疗中展现出巨大潜力。然而, 其在抗体递送效率和免疫原性方面的挑战仍需进一步解决, 以推动该技术的临床转化和广泛应用。
2.1.3 PA200
蛋白酶体激活因子PA200蛋白(PA200protein, PA200)作为单链非ATP依赖性核蛋白酶体调节组分, 通过与20S核心颗粒结合形成功能性蛋白酶体复合物。其介导乙酰化组蛋白降解的分子机制包含两个关键步骤: 首先, PA200通过其N端溴结构域类似物(bromodomain-like, BRDL)特异性识别并结合乙酰化(acetylation, AC)修饰的组蛋白; 随后, 其C端YYA基序与20S蛋白酶体α亚基相互作用, 诱导α环构象重排以开放底物通道, 促进乙酰化组蛋白进入蛋白酶体降解腔室(图3)。
PA200介导的蛋白降解机制具有独特的泛素非依赖性与ATP非依赖性的特征。区别于传统蛋白酶体途径中泛素化修饰与ATP水解供能的双重依赖性, 该通路通过乙酰化修饰的直接识别实现底物靶向, 规避了E3泛素连接酶介导的泛素标记过程及ATP驱动的底物去折叠步骤, 有效避免了E3泛素连接酶种类限制和非特异性降解致病蛋白带来的脱靶毒性等问题, 同时为能量不足的细胞降解靶蛋白提供了途径。
上述机制使其在DNA修复和精子发生中发挥关键作用, 也为治疗亨廷顿病提供创新性思路。①乙酰化组蛋白的降解在细胞受到复制应激后(如紫外线照射引起的DNA损伤)修复DNA的过程中起关键作用。而PA200-20S蛋白酶体复合物可以将乙酰化组蛋白被降解, 且使用PYR-41抑制泛素激活酶并不能阻止乙酰化组蛋白的减少, 这证明了此处降解乙酰化组蛋白是通过前文所述的非泛素依赖机制。②PA200在精子发生过程中可以时序性清除核心组蛋白。通过对比野生型小鼠和PA200敲除小鼠中组蛋白H2B(histone h2b, H2B)组蛋白H3(histone h3, H3)含量的动态变化, 可以得出结论: 在小鼠精子发生模型中, PA200促进了早期的细长精子细胞中核心组蛋白H2B和H3的降解。③PA200通过增强蛋白酶体的活性, 促进非聚集的可溶性亨廷顿蛋白N端(N terminal Huntingtin, N-Htt)在体外通过蛋白酶体途径降解, 降低细胞毒性, 为治疗亨廷顿病提供一种可能的创新治疗策略。
PA200介导的蛋白降解技术面临功能局限性, 其核心瓶颈在于底物选择性的严格修饰依赖性。该机制通过乙酰化修饰依赖型识别模式实现靶蛋白的识别, 导致其对非乙酰化修饰蛋白的降解效率显著降低。这种修饰特异性限制使其应用范围局限于特定乙酰化相关病理过程, 制约了其治疗应用的普适性。此外, PA200在不同物种中的功能相似性仍然存疑, 其在人体内精子发生的具体机制仍需进一步研究。未来研究中, 可在接近人类的哺乳动物中进行实验, 探索PA200在不同物种中是否有相似性, 可否通过其在其他物种中的精子发生机制推测其在人体内精子发生的具体机制。
2.2 溶酶体介导的降解途径
2.2.1 分子伴侣介导的自噬
分子伴侣介导的自噬(chaperone-mediated autophagy, CMA)作为选择性自噬的关键亚型, 在恶性肿瘤微环境中呈现持续活化的特征。分子伴侣CMA的作用机制分为两步。首先进行底物识别, 由HSC70对底物蛋白羧基端KFERQ样基序或其变体的胞质蛋白特异性识别, 再由HSP40、CHIP和HOP协助HSC70的底物结合, 形成HSC70复体。随后, 底物HSC70复体与溶酶体膜上的LAMP2A(溶酶体相关膜蛋白2A型)结合, 诱导LAMP2A形成多聚体通道, 同时溶酶体腔内的HSP90α协助底物蛋白去折叠并完全进入溶酶体(图4)。
CMA具有时空动态调控特征。Cuervo等构建了KFERQ-Dendra2融合荧光报告系统并建立转基因小鼠模型, 通过组织特异性启动子实现报告蛋白的定点表达, 紫外线激活Dendra2持续追踪荧光的衰减速率, 在体内动态测量CMA活性。实验数据显示: ①KFERQ-Dendra在肾脏(已知具有高基础CMA活性)、胰腺、心脏和白色脂肪组织(white adipose tissue, WAT)等组织中具有较高的降解率; ②小鼠器官之间对饥饿处理的反应存在差异, 肾脏和WAT是反应最灵敏的组织, 这些组织的CMA可能优先响应能量应激, 或具有更高的调控灵活性。此外, Dong等的实验数据显示, 部分器官存在CAM活性昼夜节律, 例如肝脏CMA活性在夜间(小鼠活动期)达峰值。
得益于高效且只针对特定序列的靶向降解机制, CMA在疾病治疗领域展现以下潜力: ①溶酶体膜直接转运机制实现蛋白质的靶向降解, 规避自噬体形成过程; ②单分子降解模式显著降低能量消耗; ③最小化膜结构重塑的能量需求; ④可针对性降解致病蛋白用于治疗神经退行性疾病, 代谢疾病和癌症, 例如帕金森病、阿尔茨海默病等; ⑤仅降解含KFERQ样基序的蛋白, 避免非特异性大量吞噬造成的细胞正常功能受损等不良反应。
尽管表现出了相当高的疾病治疗潜力, CMA在临床应用层面仍面临以下限制: ①由于溶酶体LAMP2A受体稳定性随衰老进程降低, 活性随年龄增长显著下降; ②CMA在肝脏和脑组织的递送效率差异达100倍, 面临组织特异性递送难题; ③大分子聚集体抑制CMA活性, 突变体α-synuclein(A53T和A30P, 约为300kDa的聚集体)附着于溶酶体膜并阻碍LAMP2A多聚化, 并使CMA活性下降。Kaushik等通过体外重建实验进一步证明>250kDa聚集体可完全阻断LAMP2A通道; ④CMA在正常生理条件下活性有限, 由于HSC分子的分配竞争机制, CMA被其他折叠或降解途径占用, 难以实现高效降解。
当前对CMA的分子机制研究仍存在认知空白: 其核心调控网络聚焦于溶酶体膜LAMP2A受体的多聚化过程, 但在细胞层面, 已明确的调控通路仍局限于有限分子节点, 尤其是在考虑存在的CMA激活刺激的多样性时。此外, 在不表达LAMP2A的物种中, 内体微自噬是否是CMA替代品, 或这些物种中是否可能存在另一种CMA等效途径, 仍需要更多探索为溶酶体膜两侧伴侣的能量需求、驱动力和作用方式提供新的见解。
2.2.2 基于双特异性抗体的蛋白靶向嵌合体技术
自2001年Sakamoto等开创性提出PROTACs技术以来, 该领域历经20年发展已形成突破性优势。然而, 其技术同样存在局限: 由于泛素介导的降解途径发生在细胞内, 分子PROTACs目前主要攻击细胞内的靶点, 许多膜蛋白无法被PROTACs技术靶向, 因为这类蛋白在胞内段并无合适的结合口袋供小分子配体结合, 需要一种降解细胞表面蛋白的新策略来扩大靶向降解蛋白范围。
基于双特异性抗体的蛋白靶向嵌合体技术(antibody-based PROTACs, AbTACs)通过双特异性抗体的结构框架, 整合PROTACs技术核心原理, 形成新型降解平台。AbTACs通过双特异性抗体, 一个臂识别靶蛋白, 另一个臂连接E3泛素连接酶(如RNF43), 从而可以将E3泛素连接酶拉到靶蛋白周围。与传统的PROTACs不同, AbTACs不直接进入细胞, 而是通过结合细胞表面受体和膜相关E3泛素连接酶(如RNF43、ZNRF3等)来诱导邻近蛋白质的内吞和降解(图5)。Cotton等以AbTACs通过招募膜结合E3泛素连接酶RNF43来降解PD-L1为模型研究AbTACs的作用机制。PD-L1有一个31个氨基酸长的胞质域, 但没有已知能够与该结构域结合的小分子配体, 这使得用传统小分子PROTACs来靶向PD-L1具有挑战性。而在构建的PD-L1靶向模型中, AbTACs同时结合细胞膜上的PD-L1和RNF43, 形成三元复合物, RNF43胞内RING结构域被激活, 暴露其E3泛素连接酶活性中心, PD-L1在泛素化后进而内化并被溶酶体降解, E3泛素连接酶可再生后循环利用。实验结果进一步显示AbTACs以依赖RNF43和溶酶体的方式降解细胞中PD-L1。
AbTACs在药代动力学层面展现出显著优势: 其血浆半衰期可达数天量级, 单次给药即可维持长效治疗效果, 这得益于抗体分子的长半衰期特性与E3泛素连接酶的可循环利用机制。然而, AbTACs技术在临床应用层面面临双重挑战: ①免疫原性风险显著, 工程化抗体框架可能引发宿主ADA反应; ②大分子质量特征(>100kDa)导致实体瘤渗透效率受限。对此, 当前解决方案包括: 采用脂质纳米颗粒或金属有机框架纳米载体提升递送效率, 结合超声微泡介导的递送系统增强肿瘤穿透性等。
此外, AbTACs结构依然可以在以下方面做进一步优化以增加蛋白质降解效率: ①E3泛素连接酶和E3泛素连接酶结构域的水平; ②AbTACs结合臂的化合价、柔韧性和取向; ③不同跨膜E3泛素连接酶对细胞的依赖性。AbTACs未来可研究应用于靶向更复杂的细胞表面蛋白, 例如多通道跨膜受体, 为精准医疗时代提供了强有力的工具。
此外, 为提升AbTACs的靶向性, 研究人员开发了光控AbTACs技术, 通过整合光响应分子开关实现蛋白质降解的时空精准调控。Liu等在传统AbTACs基础上引入光激活机制, 使蛋白降解仅发生在特定光照区域, 从而增强肿瘤治疗的靶向性并减少脱靶效应。该技术的核心设计策略在于: ①光响应调控, 利用光敏基团(如光笼分子)控制AbTACs的活性, 实现按需降解; ②光照激活后, 仅在肿瘤部位触发靶蛋白降解, 避免全身性不良反应。同时, 光控AbTACs具有降低免疫原性的附加优势: 光笼化状态可隐藏抗体表位, 减少宿主免疫系统识别风险。
3展望与挑战
本文系统分析了基于生物大分子的靶向蛋白降解药物, 重点探讨了基于泛素−蛋白酶体系统和溶酶体途径的多种技术路线, 包括bioPROTACs、TRIM-Away、PA200、CMA和AbTACs等。这些技术通过利用生物大分子的高特异性和多功能性, 克服了传统小分子降解剂的局限性, 展现了在精准靶向治疗中的巨大潜力。然而, 这些技术仍面临以下挑战: ①递送效率低下: 以bioPROTACs、TRIM Away和AbTACs为例, 大分子药物(如多肽/抗体)因分子质量过大难以直接穿透细胞膜, 需依赖低效的内吞作用或外源递送系统(如病毒载体)进入细胞。此外, 血脑屏障等生理屏障进一步阻碍药物对中枢神经系统靶点的有效递送, 显著降低降解效率; ②降解技术靶向特异性仍存在提升空间; ③bioPROTACs、TRIM-Away和AbTACs均因依赖外源蛋白或抗体成分而面临显著的免疫原性风险, 这一共性问题可能限制药物用于人体长期临床应用。例如, bioPROTACs通常包含细菌或病毒来源的E3泛素连接酶结合肽或人工设计的多肽, 这些成分可能被免疫系统识别并触发ADA反应的产生, 导致药物中和及过敏反应。为了更系统地总结基于生物大分子的靶向蛋白降解药物的研究进展与挑战, 表2对本文所提及技术的优势、挑战以及常见作用位点进行了总结。
因此, 未来生物大分子靶向蛋白药物降解技术的发展方向主要分为以下几个方向: ①针对大分子跨膜效率低的问题, 开发脂质纳米颗粒、病毒仿生载体等新型递送系统, 提升药物穿透细胞膜及血脑屏障的能力, 同时能够实现靶向蛋白降解药物的体内靶向递送, 降低药物的脱靶毒性; ②提高靶向蛋白降解药物的特异性响应, 例如光控、pH响应和小分子诱导的降解剂, 或进一步设计组织特异性启动子, 从而实现生物大分子靶向蛋白降解药物的特异性作用; ③降低生物大分子药物的免疫原性, 可采用减少异源蛋白序列的使用或通过化学修饰mRNA实现细胞内bioPROTAC表达, 也可通过局部递送药物减少药物系统性分布, 从而降低免疫激活风险; ④结合生物信息学和人工智能算法, 预测靶蛋白及生物大分子药物结构, 基于结构合理设计优化双功能分子(bioPROTACs)。或基于生物信息学分析E3泛素连接酶的组织及细胞特异性丰度差异, 合理利用特定泛素连接酶, 从而提高降解效率。同时, 生物大分子存在自身泛素化降解的可能性, 以及与靶蛋白的结合存在蛋白−蛋白界面, 可能会占据并影响靶蛋白的泛素化修饰位点, 因此采用分子动力学模拟及空间结构预测对于生物大分子的靶向蛋白降解药物的开发尤为重要。随着对生物大分子靶向蛋白药物降解技术的深入研究和不断优化, 以及人工智能技术在大分子药物结构预测和辅助设计中的应用拓展, 未来基于大分子的靶向蛋白降解药物将能针对更多药物靶点实现高效特异性降解, 为不同疾病的临床治疗提供新的思路。

来源:Internet


