您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-09-08 21:18
本文包含研发不同阶段的GMP和质量体系要求,包括以下质量体系项目在不同阶段如何实施:
Quality management/ oversight
质量管理/监督
Personnel training
人员培训
Documentation & records
文件记录
Product release
产品放行
Change management
变更管理
Deviations/ investigations
偏差/调查
CAPA (Corrective Action Preventive Action)
CAPA
Auditing
审计
Quality agreements
质量协议
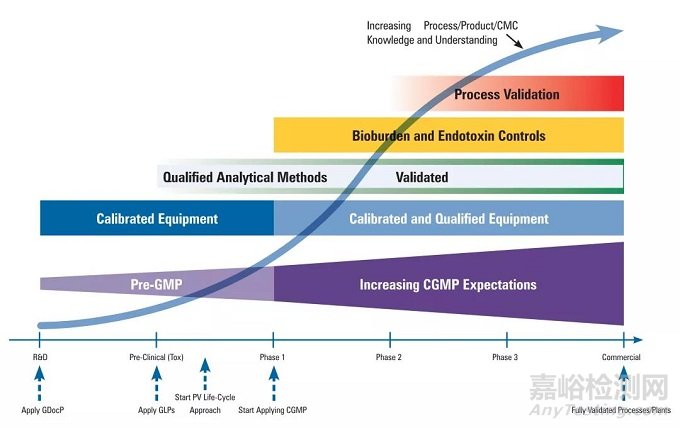
研发不同阶段的GMP和质量体系要求
R&D阶段
Personnel have science background & aretrained in routine laboratory practices but should also be made aware of GMPprinciples.
人员有科学背景并接受日常实验室操作的培训且应使其了解GMP原则
Signed notebook records are kept ofproduction and testing activities. If batches fail, they are studied toincrease product and process knowledge.
应保存生产和检验活动的签字记录。如果批次失败,对其进行研究以增加产品和工艺知识。
R&D activities are well documented inthe note books, as well as in periodic development reports.
研发活动在记录本和定期开发报告中都有很好的记录。
There is a need to document source/pedigree/chain of custody of biological starting materials – cell ancestor andany animal materials used to create the initial cell line to the extentfeasible (Reference Section 6.0 and Section 3.3.2 of this technical report formore information).
有必要记录生物起始物料(用于创建初始细胞系的细胞供体和任何动物材料)的来源/谱系/保管链。
Toxicity Studies
毒性研究阶段
GLP practices are implemented as perregulations in specific global regions
GLP实践是按照全球特定地区的法规实施的
EU and FDA GLP Requirements cover the areasof:
欧盟和FDA GLP要求涵盖以下领域:
Organization and Personnel
组织和人员
Facilities
设施
Equipment
设备
Facility Operation
设施操作
Articles
物品
Protocol and Conduct
方案和执行
Records and Reports
记录和报告
Disqualification
不合格
There is a need to document the Tox lot andreference standard material with sufficient diligence to allow comparability toclinical/GMP lots later
有必要对毒理学批次和参考标准进行充分的记录,以便日后与临床/GMP批次进行比较
Phase 1
一期阶段
It is expected that a laboratory directorwith a science background is in charge of the Quality Unit (or equivalentfunction) and reviews all procedures and documents.
要求有科学背景的实验室负责人负责质量部门(或同等职能)并审查所有程序和文件。
Use of batch records are highly recommendedbut could be generic.
强烈建议使用批记录,但可以是通用的。
The Bulk Drug Substance is released byQA/QP after satisfactory review of the manufacturing and analytical records anddata (e.g., Completed batch record, analytical results, COA, environmental andwater monitoring data, deviations and changes), as well as compliance to theinvestigational new drug registration (e.g., IND, IMPD).
在对生产和检验记录和数据(如完整的批记录、检验结果、COA、环境和水监测数据、偏差和变更)以及临床试验新药注册(如IND、IMPD)的符合性进行满意审查后,散装药用物质由QA/QP放行。
Manufacturing or testing deviations orunexpected events that do not impact product quality or patient safety shouldbe documented, but could be appended to the executed batch records. Formaldeviation and CAPA systems are recommended, albeit a simple and uncomplicatedsystem, with the level of investigation dictated by the severity of theincident.
应记录不影响产品质量或患者安全的生产或检验偏差或意外事件,但可添加在已执行的批记录中。建议使用正式的偏差和CAPA系统,即便是一个简单和不复杂的系统,调查级别由事件的严重程度决定。
Change management is important duringdevelopment of a product and the process to catalogues changes and facilitatesproduct /process understanding. A system should be in place at all GMPmanufacturing facilities.
变更管理在产品开发和对变更进行分类并促进产品/工艺理解的过程中非常重要。在所有GMP生产设施都应有一个系统。
Auditing/self-assessment is recommended,even though it is not mandated.
建议进行审计/自检,尽管这不是强制性的。
Testing or manufacturing conducted by athird party should be subject to a written agreement that might outline criticalquality expectations but a separate quality agreement may not be required.
由第三方进行的检验或生产应遵守一份书面协议,说明关键的质量期望,但单独的质量协议则可能不需要。
Phase 2 →Phase 3
二期→三期阶段
Responsibilities are governed by CGMP(e.g., ICH Q7 Eudralex - Volume 4 Good Manufacturing Practice Guidelines, andits Annex 13 by phase of development for some items (e.g., as methods are fullyvalidated or transferred, as master batch records are created). QA/QPresponsibilities must not be delegated to another functional area (unlessallowable by local law), but may be contracted to a qualified external serviceprovider.
职责由CGMP(例如,ICH Q7Eudralex - Volume 4 Good Manufacturing Practice Guidelines,及其附件13)管理,按某些项目的开发阶段(例如,方法完全验证或转移时,主批记录创建时)。QA) / QP的职责不能委托给其他职能部门(除非当地法律允许),但可以委托给有资质的外部服务提供商。
QA/QP takes a more active role in directinginvestigations and approving findings and CAPAs.
QA/ QP在管理调查、审批结果和CAPA方面发挥更积极的作用。
The reporting structure and hierarchy ofthe Quality Unit should ensure its ability to be independent from production.(From ICH Q7: There should be a quality unit(s) that is independent ofproduction and that fulfills both quality assurance (QA) and quality control(QC) responsibilities).
质量部门的报告结构和等级应确保其可以独立于生产。(来自ICH Q7:应该有一个独立于生产并同时履行质量保证(QA)和质量控制(QC)职责的质量部门)。
Quality standards (e.g., policies, SOPs)must be reviewed and approved by QA. Even when these standards and procedureshave not been formally changed, they should be subject to periodic review inorder to ensure that they are still valid and up to date with actual practices.It is recommended that for each phase of clinical development, the relevantsummary development reports should be completed to review process
development activities and results. Thereports should include an evaluation of deviations and unexpected results thatare encountered during clinical production, scale up, tech transfer, characterizationstudies, etc.
质量标准(如方针、标准操作规程)必须由质量保证部门(QA)审核和批准。即使这些标准和程序没有正式变化,也应定期审查,以确保它们仍然有效,并与实际做法保持一致。建议对于临床开发的每个阶段,都应完成相关的总结开发报告,以审查工艺开发活动和结果。报告应包括在临床生产、放大、技术转移、特征研究等过程中遇到的偏差和意外结果的评估。
The Bulk Drug Substance is released by QA/QPafter review of the completed batch record, COA, environmental and watermonitoring data, deviations and changes, the investigational new drugregistration (e.g., IND, IMPD), and any other relevant information available inthe product specification file as specified in the procedures for batchrelease. QA/QP can delegate the release of manufactured intermediates to otherqualified personnel upon formalized agreements and acceptance.
散装药物物质在完成对完整批记录、COA,环境和水的监测数据,偏差和变更,临床试验新药注册(例如,IND,IMPD),以及批放行程序中指定的产品标准文件中规定的其他任何相关信息的审核后,由QA / QP放行。在正式协议和任命后,QA/QP可以将所生产中间体的放行委托给其他有资质的人员。
As clinical development proceeds, moredetailed batch records with acceptance criteria or target values should bedeveloped. Master batch records should be used prior to conducting processvalidation. Deviations should be recorded in the batch records as well asrequire formal investigations and CAPA. Deviation and investigations areincreasingly thorough as clinical development proceeds. By Phase 3 a formaldeviation tracking system and a CAPA system should be in place.
随着临床开发的进行,应该指定更为详细的带有接受标准或目标值的批记录。主批记录应在进行工艺验证之前使用。偏差应在批记录中记录,并要求正式调查和CAPA。随着临床开发的进行,偏差和调查越来越彻底。在第三阶段,正式的偏差跟踪系统和CAPA系统应到位。
Clinical materials should not bedistributed to external partners of the clinical supply chain until all opendeviations, test results, or other documentation are closed and approved byQA/QP.
在所有偏差、检测结果或其他文件关闭并经QA/QP批准之前,不应将临床物料分发给临床供应链的外部合作伙伴。
Changes during the initial phases ofclinical development should be documented and justified based on the magnitudeof the change. Significant changes to the process should be conducted inaccordance with a written change management procedure and the IND should beupdated accordingly. As clinical development progresses a formal change controlprogram should be developed for Phase 3. Any potential impact of the change onon-going trials should be considered. Changes made during production operationsmust be reviewed by QA/QP during batch disposition.
在临床开发开始阶段的变更应进行记录,并根据变更的大小进行论证。工艺的重大变更应按照书面变更管理程序进行,IND也应相应更新。随着临床开发的进展,应该为第三阶段制定一个正式的变更控制程序。变更对正在进行的试验的任何潜在影响都应予以考虑。在生产操作过程中所做的变更必须在批处理过程中由QA/QP进行审核。
QA/QP audits should be conducted based upona risk assessment. Typically, fewer audits are conducted for early phasedevelopment operations and compounds.
应基于风险评估进行QA/QP 审计。通常,对开发早期阶段的操作和合成进行的审计较少。

来源:GMP


