您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-08-23 12:48
医疗器械网络安全的话题越来越热门,而且现在各个国家监管机构都已经很重视医疗器械网络安全的合规性,但是面对众多的网络安全要求,可能还有很多企业有点无从下手,不知道哪些文件是必须要学习的。下面小编就整理了关于医疗器械网络安全入门级的指导原则或者标准,供大家参考学习。
基本概念
HIPAA(Health Insurance Portability and Accountability Act)
健康保险可携带与责任法案
美国HIPAA出台之前,医疗卫生行业针对保护患者健康信息没有通用的要求或安全标准。由于无规可循,医疗服务提供商不能自信地断言病人敏感数据得到了适当保护。但在“健康保险可携带与责任法案”(HIPAA)经联邦政府审议通过之后,医疗服务提供商便有了需要遵循的明确标准和要求,以确保妥善地保存、转移和传输病例和病患信息。
安全规则需要适用于以电子形式接收、传输或存储的可单独识别的健康信息(称为ePHI)包括:
- 隐私规则
- 交易规则和代码集
- 唯一标识符规则(国家提供者标识符)
- 执行规则等等
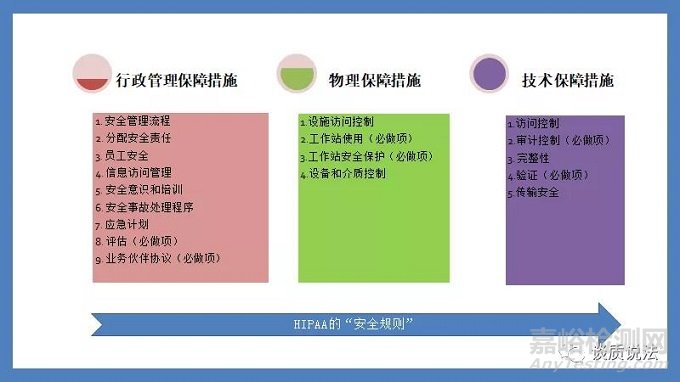
HIPAA的“安全规则”针对其所涉及到的实体规定了保障措施,包括行政管理保障措施(Administrative Safeguards)、物理保障措施(PhysicalSafeguards)和技术保障措施(Technical Safeguards)。
Cybersecurity信息安全
信息安全-是防止未经授权的访问、修改、误用或拒绝的过程使用或未经授权使用存储、访问或从数据库传输的信息向外部接受者提供医疗器械。
医疗器械网络安全是指保持医疗器械相关数据的保密性(confidentiality)、完整性(integrity)和可得性(availability)
保密性:指数据不能被未授权的个人、实体利用或知悉的特性,即医疗器械相关数据仅可由授权用户在授权时间以授权方式进行访问;
完整性:指保护数据准确和完整的特性,即医疗器械相关数据是准确和完整的,且未被篡改;
可得性:指根据授权个人、实体的要求可访问和使用的特性,即医疗器械相关数据能以预期方式适时进行访问和使用。
另外,医疗器械网络安全特性还包括真实性(authenticity)、可核查性(accountability)、抗抵赖(non-repudiation)和可靠性(reliability)等。
见GB/T 29246-2012。医疗器械的网络安全应当符合相应法律法规和部门规章的要求。
医疗器械产品的网络安全特性
应当基于医疗器械相关数据的类型、功能、用途、交换方式及要求,并结合医疗器械产品的预期用途、使用环境和核心功能以及预期相连设备或系统(如其它医疗器械、信息技术设备)的情况来确定医疗器械产品的网络安全特性,并采用基于风险管理的方法来保证医疗器械产品的网络安全:识别资产(asset,对个人或组织有价值的任何东西)、威胁(threat,可能导致对个人或组织产生损害的非预期事件发生的潜在原因)和脆弱性(vulnerability,可能会被威胁所利用的资产或风险控制措施的弱点),评估威胁和脆弱性对于医疗器械产品和患者的影响以及被利用的可能性,确定风险水平并采取适宜的风险控制措施,基于风险接受准则评估剩余风险。
(一)数据考量(数据类型及数据交换方式)
医疗器械相关数据从内容上可分为2种类型:
健康数据:标明生理、心理健康状况的私人数据(“Private Data”,又称个人数据“Personal Data”、敏感数据“Sensitive Data”,指可用于人员身份识别的相关信息),涉及患者隐私信息;
设备数据:描述设备运行状况的数据,用于监视、控制设备运行或用于设备的维护保养,本身不涉及患者隐私信息。
医疗器械相关数据的交换方式可分为以下两种情况:
网络:通过网络(包括无线网络、有线网络)进行电子数据交换或远程控制,需要考虑网络相关要求(如接口、带宽等),数据传输协议需考虑是否为标准协议(即业内公认标准所规范的协议),远程控制需考虑是否为实时控制;
存储媒介:通过存储媒介(如光盘、移动硬盘、U盘等)进行电子数据交换,数据储存格式需考虑是否为标准格式(即业内公认标准所规范的格式)。
(二)技术考量
用户访问控制机制应当与医疗器械产品特性相适应,
包括但不限于用户身份鉴别方法(如用户名、口令等)、用户类型及权限(如系统管理员、普通用户、设备维护人员等)、口令强度设置、软件更新授权等。
医疗器械相关数据在网络传输或数据交换过程中应当保证保密性和完整性,同时平衡可得性的要求,特别是具有远程控制功能的医疗器械。鉴于预期用途、使用环境的限制,医疗器械对于网络安全威胁的探测、响应和恢复能力应当与医疗器械的产品特性相适应。
可采用加密、数字签名、标准协议、校验等技术来保证医疗器械的网络安全。
可采用防火墙、入侵检测和恶意代码防护等技术来保证医疗器械的网络安全。
医疗器械网络安全能力建设可参照相关的国际、国家标准和技术报告,如IEC/TR 80001-2-2规范了十九项网络安全能力:自动注销(ALOF)、审核控制(AUDT)、授权(AUTH)、安全特性配置(CNFS)、网络安全产品升级(CSUP)、健康数据身份信息去除(DIDT)、数据备份与灾难恢复(DTBK)、紧急访问(EMRG)、健康数据完整性与真实性(IGAU)、恶意软件探测与防护(MLDP)、网络节点鉴别(NAUT)、人员鉴别(PAUT)、物理锁(PLOK)、第三方组件维护计划(RDMP)、系统与应用软件硬化(SAHD)、安全指导(SGUD)、健康数据存储保密性(STCF)、传输保密性(TXCF)和传输完整性(TXIG)。
可根据医疗器械的产品特性考虑其网络安全能力要求的适用性。
各个监管机构对于网络安全的要求。
FDA的要求:
美国食品与药物管理局在官网上详细列举了其近年来改进医疗设备网络安全的相关措施,原因是FDA发现各类医疗设备既存在一定收益,也都带来多种风险。对于美国食品与药物管理局(简称FDA)而言,当能够以合理方式确保受众收益超过风险时,其即允许厂商将相关设备投放市场。如今,医疗设备正越来越多地与互联网、医院网络以及其它医疗设备对接,旨在改善医疗保健效能并增强医疗服务机构为患者提供的诊疗能力。但在交付这些能力的同时,此类设备也将增强网络安全威胁水平;与其它计算机系统一样,医疗设备可能受到安全漏洞的影响,进而导致设备安全性与有效性遭到破坏。
FDA准备和实施了很多举措,具体包括:
2017年5月18日至19日,FDA与国家科学基金会(简称NSF)以及国土安全部科学技术部(简称DHS,S&T)合作举办公共研讨会,题为《医疗器械网络安全:监管科学差距分析》。此次研讨会的目标在于研究FDA目前正在参与的新兴研究机遇; 促进利益相关方合作以确保监管科学挑战; 讨论应对这些挑战的对应创新战略; 同时鼓励各利益相关方社区积极开发分析工具、流程与最佳实践,从而提升医疗设备的安全水平。
2017年1月12日,FDA就《医疗器械上市后网络安全管理》指南研讨会,邀请各利益相关方出席以了解指导内容并提出相关疑问。此份指南的最终版本发布于2016年12月27日,其中向各制造商通报了FDA方面就医疗设备上市后,立足产品整体生命周期之内的市场营销与分销流程,提出的网络安全漏洞结构化及全面管理方法层面的相关建议。
2018年10月18日,FDA发布《医疗器械上市前网络安全管理》规定。此份指南性草案向行业提供关于网络安全设备设计、标注以及FDA为可能存在网络安全风险的设备在上市前申报文件中需要体现的建议性内容。
2018年10月,FDA为MITRE的《医疗设备网络安全区域性事件准备与响应手册》提供指导性支持。此份手册描述了准备事务的具体类型,旨在帮助各医疗保健服务组织(简称HDO)更好地为涉及医疗设备的网络安全事件做好准备,并为产品开发人员提供足以解决大规模、影响广泛的安全事件的必要方法,最终避免事件对患者人身安全造成影响。
除此之外,FDA还与多家利益相关方签订了两份新的谅解备忘录,计划建立起MedISAO与Sensato-ISAO两大信息共享分析组织(简称ISAO)。这些共享分析组织的目标,在于为制造商提供与FDA共享关于潜在安全漏洞与新兴威胁信息的机会,并通过尽早解决这些问题以帮助制造商更好地保护患者安全。
《医疗器械上市前网络安全管理》General Principles一般原则
Address cybersecurity during design &development 在设计和开发过程中解决网络安全问题
Identification of assets, threats, and vulnerabilities 识别资产、威胁和漏洞
Consider type & likelihoodvulnerabilities may result in patient harm 考虑漏洞的类型和可能性,以及可能导致病人的伤害
Assess the likelihood of exploit 评估伤害产生的可能性
Assess residual risk, risk acceptance 评估剩余风险,风险接受
Consider device intended use and useenvironment考虑设备的预期用途和使用环境
CybersecurityFunctions网络安全功能:
Security controls appropriate for datainterfaces 适合于数据接口的安全控制
Balance security safeguards and usability 平衡安全保障和可用性
Security controls appropriate for intended 适合于目标用户的安全控制
Security controls should not hinderintended use during emergency situations 安全控制不应妨碍在紧急情况下的预期使用
Detail security controls chosen for device 详细的安全控制选择设备
Justification for security controls chosenor not chosen 选择或不选择安全控制的理由
FDA把网络安全风险归为两个类别:
Medical devices capable of connecting(wirelessly or hard-wired) to another device, to the Internet or other network,or to portable media (e.g. USB or CD) are more vulnerable to cybersecuritythreats than devices that are not connected.
• Tier 1 “Higher Cybersecurity Risk”
A device is a Tier 1 device if thefollowing criteria are met:
1)The device is capable of connecting(e.g., wired, wirelessly) to another medical or non-medical product, or to anetwork, or to the Internet; AND
1类是指该设备能够连接(如有线、无线)到另一医疗或非医疗产品、网络或Internet;和
2)A cybersecurity incident affecting thedevice could directly result in patient harm to multiple patients. 影响设备的网络安全事件能直接导致对多名患者的伤害植入式心律转复除颤器(ICDs)、起搏器、左心室辅助设备(LVADs)、大脑刺激器和神经刺激器、透析设备、 输液和胰岛素泵,以及与这些设备交互的支持连接系统,如家庭监测器,以及具有命令和控制功能的系统
• Tier 2 “Standard Cybersecurity Risk”
• A medical device for which the criteria fora Tier 1 device are not met.其它的设备都是2类。
《医疗器械上市后网络安全管理》:FDA Post-Market Guidance
• Applies to any marketed and distributedmedical device including 适用于任何市场销售和分布式医疗设备,包括:
– Medical devices that containsoftware, firmware, or programmable logic
包含软 件、固件或可编程逻辑的医疗设备
– Software that is a medicaldevice, including mobile medical applications医疗设备软件,包括移动医疗应用程序
– Medical devices that areconsidered part of an interoperable system 被认为是可互操作系统的一部分的医疗设备
– Legacy devices - devicesalready on the market or in use. 遗留设备 - 已经上市或正在使用的设备。
• Emphasizes that manufacturers shouldmonitor, identify, and address cybersecurity vulnerabilities and exploits aspart of their postmarket management of medical devices 强调制造商应监控、识别和解决网络安全漏洞和漏洞,作为医疗设备上市后管理的一部分
• Establishes a risk-based framework forassessing when changes to medical devices for cybersecurity vulnerabilitiesrequire reporting to the Agency 建立一个基于风险的框架,用于评估医疗设备的网络安全漏洞何时需要向机构报告
• Outlines circumstances in which FDA doesnot intend to enforce reporting requirements under 21 CFR part 806. 以概述FDA不打算在21 CFR第806部分下强制执行报告要求的情况。
• In order to demonstrate a reasonableassurance of safety and effectiveness for software devices, documentationrelated to the requirements of the Quality System Regulation (QSR) (21 CFR Part820) is often a necessary part of the premarket submission. 为了证明对软件设备的安全性和有效性的合理保证,与质量体系规范(QSR) (21 CFR Part 820)要求相关的文档通常是上市前提交的必要部分
• As part of QSR design controls, amanufacturer must “establish and maintain procedures for validating the devicesdesign,” which “shall include software validation and risk analysis, whereappropriate.” 21 CFR 820.30(g).
• 制造商必须“建立和维护验证设备设计的程序”,其中“应包括软件验证和风险分析”
• As part of the software validation and riskanalysis required by 21 CFR 820.30(g), software device manufacturers may needto establish a cybersecurity vulnerability and management approach
• 作为21 CFR 820.30(g)要求的软件验证和风险分析的一部分,软件设备制造商可能需要建立网络安全漏洞和管理方法
• FDA recommends that this approach include aset of cybersecurity design controls to ensure medical device cybersecurity andmaintain medical device safety and effectiveness.确保医疗设备的网络安全,并保持医疗设备的安全和有效性
– 21 CFR part 820: QualitySystem Regulation
– 21 CFR part 820.198: Complaint handling
– 21 CFR part 820.22: Quality audit
– 21 CFR part 820.100: CAPA
– 21 CFR part 820.30(g): Software validation and risk analysis
– 21 CFR 820.200: Servicing
欧盟要求
指导原则:MDCG2019-16 医疗器械网络安全指导原则
医疗器械协调工作组(MDCG)前期发布了“医疗器械信息安全指南, 2019年12月”。该指南不具有法律约束力,但公告机构必须遵循。这意味着您作为制造商也需要遵循。不幸的是,该指南并未就如何处理该问题提出明确的框架,而是概述了也可在其他资源中找到的要求和方法,例如:
uISO /IEC 80001-1风险管理在包含医疗器械的IT网络中的应用
uIEC /TR 80001-2-2风险管理在包含医疗器械的IT网络中的应用第2-2部分:医疗器械安保需求、风险和控制的披露和沟通指南。
uAAMITIR 57医疗器械安保原则-风险管理
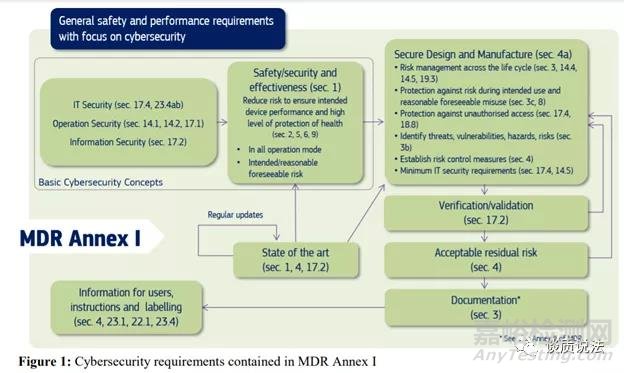
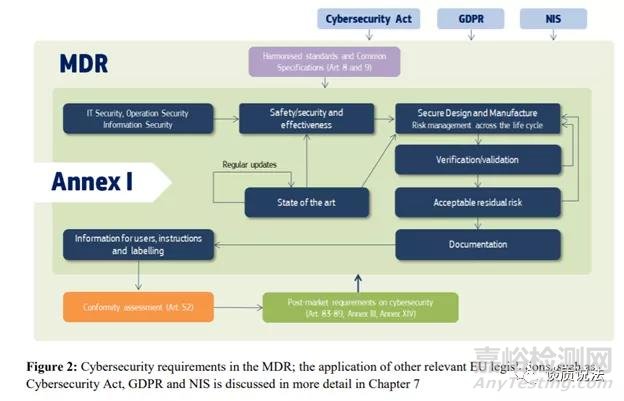
MDCG指南把MDR一般安全和性能要求(GSPR)关联了起来。介绍了深度防御,良好网络安全防范 (FDA指导中的基本安全防范)以及网络安全风险与产品安全风险之间的关系这样一些概念。
还引入可用性工程与网络安全之间的联系:脆弱性被视为合理可预见的滥用的促成因素。
MDCG指南提出了6个最佳实践,主要使用了ISO 13485和IEC 62304中的设计和维护过程中步骤:
1 管理上的安全
-ISO13485 4.1,用于安全风险管理过程;
-IEC62304 5.1:软件开发计划;
-IEC62304 6.1:软件维护
2 安全要求规范
-IEC62304 5.2:软件需求分析
3 通过设计确保安全,包括深度防御
-IEC62304 5.3:软件体系结构;
4安全实施
-IEC62304 5.4:软件详细设计;
-IEC62304 5.5:软件实施和单元验证;
-以及SOUP管理的正确性
5安全验证和确认
-IEC62304 5.6:软件集成测试;
-IEC62304 5.7:软件系统验证;
6安全相关问题的管理
-IEC62304 6.2:问题和修改分析;
-IEC62304 9:问题解决
7安全更新管理
-IEC 623046.3:修改实施;
-IEC62304 8.2:变更控制;
8 安全准则
-5.8软件发布;
-以及软件文档,请参阅IEC 82304-1第7节。
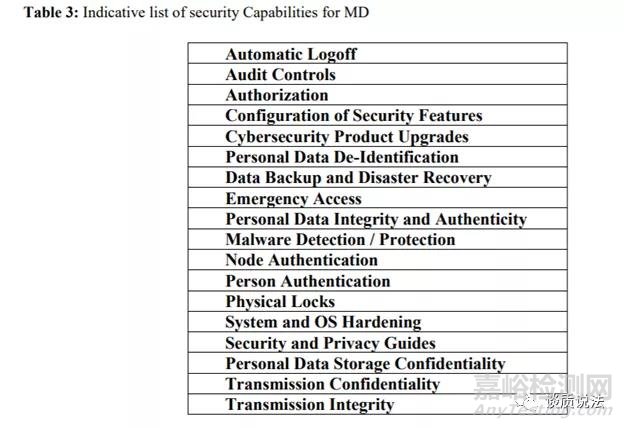
指南也提到验证与确认
安全验证和确认测试的主要手段还是测试,方法可以包括安全功能测试、模糊测试,漏洞扫描和渗透测试。额外的安全测试可以通过使用安全的代码分析工具和工具,扫描开放源代码和库中使用的产品,确定组件与已知问题。
指南也描述了关于说明书,PMS和警戒等内容。
彩蛋:如果大家有时间,建议仔细研究2020.4.20,IMDRF就医疗器械网络安全发布的官方指南——《医疗器械网络安全原则与实践》(Principles and Practicesfor Medical DeviceCybersecurity),这个在全球监管协调方面具有重要且特别的意义。
中国NMPA要求
目前我们主要还是参考《医疗器械网络安全注册技术审查指导原则》,这个原则适用于医疗器械网络安全,包括具有网络连接功能以进行电子数据交换或远程控制的第二类、第三类医疗器械产品,其中网络包括无线、有线网络,电子数据交换包括单向、双向数据传输,远程控制包括实时、非实时控制。也适用于采用存储媒介以进行电子数据交换的第二类、第三类医疗器械产品,其中存储媒介包括但不限于光盘、移动硬盘和U盘。
医疗器械网络安全是指保持医疗器械相关数据的保密性(confidentiality)、完整性(integrity)和可得性(availability).
通过之前的介绍,相信大家对于医疗器械网络安全的基本概念和各监管机构的要求已经有了初步的认知。接下来,请同小编一起实践探讨准备网络安全文档。
(一)基本考量
网络安全更新(包括自主开发软件和现成软件)根据其对医疗器械的影响程度可分为以下两类:
1.重大网络安全更新:影响到医疗器械的安全性或有效性的网络安全更新;
2.轻微网络安全更新:不影响医疗器械的安全性与有效性的网络安全更新,如常规安全补丁。
(二)网络安全描述文档
1.基本信息
描述医疗器械产品的相关信息:
(1)类型:健康数据、设备数据;
(2)功能:电子数据交换(单向、双向)、远程控制(实时、非实时);
(3)用途:如临床应用、设备维护等;
(4)交换方式:网络(无线网络、有线网络)及要求(如传输协议(标准、自定
义)、接口、带宽等),存储媒介(如光盘、移动硬盘、U盘等)及要求(如存储格式(标准、自定义)、容量等);对于专用无线设备(非通用信息技术设备),还应提交符合无线电管理规定的证明材料;
(5)安全软件:描述安全软件(如杀毒软件、防火墙等)的名称、型号规格、完整版本、供应商、运行环境要求;
(6)现成软件:描述现成软件(包括应用软件、系统软件、支持软件)的名称、型号规格、完整版本和供应商。
2.风险管理
提供医疗器械网络安全风险管理的分析报告和总结报告,确保全部剩余风险均是可接受的。
3.验证与确认
提供网络安全测试计划和报告,证明医疗器械产品的网络安全需求(如保密性、完整性、可得性等特性)均已得到满足。同时还应提供网络安全可追溯性分析报告,即追溯网络安全需求规范、设计规范、测试、风险管理的关系表。
对于安全软件,应提供兼容性测试报告。
对于标准传输协议或存储格式,应提供标准符合性证明材料,而对于自定义传输协议或存储格式,应提供完整性测试总结报告。
对于实时远程控制功能,应提供完整性和可得性测试报告。
4.维护计划
描述软件(含现成软件)网络安全更新的维护流程,包括更新确认和用户告知。
(三)常规安全补丁描述文档
提交软件(含现成软件)常规安全补丁的情况说明(补丁描述、影响分析、用户告知计划)、测试计划与报告、新增已知剩余缺陷情况说明(证明新增风险均是可接受的)。

来源:谈质说法


