您当前的位置:检测资讯 > 检测案例
嘉峪检测网 2019-04-28 12:00
1.前言
在药品设计过程中,辅料的选择至关重要。对于辅料及其在处方中的用量选择,不仅基于它们的功能性,更重要的是要考虑药物与辅料之间的相容性。所谓的不相容性,可以定义为药物与处方中一种或多种辅料发生不良相互作用,从而导致制剂在物理、化学、微生物学或治疗性质方面的改变。因此,辅料相容性的研究主要是用于预测药物在最终剂型中可能潜在的不相容性,同时为申报注册法规文件所需的处方中辅料及其用量的选择提供合理依据。
辅料相容性的研究通常被认为是很普通并且繁琐的。但是,这些研究恰恰是药物研发过程中非常重要的工作,原因在于:包括处方的选择、药物稳定性的评估,降解产物的鉴定,以及对于相互作用机制的了解,都可以从辅料相容性研究所获得的知识中得到有益指导。如果发现药物稳定性不能尽如人意,就要采取对策以提高其稳定性。因此,在药品开发的后阶段进行系统的,周详计划和执行的相容性研究,可以有效地节省由于稳定性问题而浪费的资源和拖延的时间;同时,如果当药物产品进入后期开发的阶段,辅料相容性的研究对于引起稳定性问题的原因推测也非常有帮助。
而且随时间的推移,监管的期望对此有了显著的增加。正如一直都在提倡的质量源于设计(QbD)的倡议那样,这种趋势将会一直延续。在进入申请的开发报告中,需要药物与辅料的相容性数据以证明对处方成分选择的正确性。基于辅料对药品和生产工艺的影响,相关法规已经越来越多地关注其关键质量特性(critical quality attributes,CQA)和控制策略。
从药物开发过程的角度而言,通常是在对药品(药物活性成分,API)液体和固体稳定性有一定的了解之后开展这些研究,但应在处方开发之前。剂型中不相容导致的变化可以归纳为以下几点
①颜色或外观的变化;
②机械性能的损失(如片剂硬度);
③溶出行为的变化;
④物理晶型的转变,
⑤升华导致的损失;
⑥药效降低;
⑦降解产物的增加。
2.药物与辅料之间的化学作用
在药物制剂中能观察到最普遍的反应为水解作用、脱水作用、同分异构化作用、消除作用、成环作用、氧化作用,光降解作用,以及与处方成分(辅料及其杂质)之间的特殊反应。而影响上述反应的主要因素为:温度、酸碱度(pH值)、固体中的水分、环境中的相对温度、催化剂的存在、光照、氧气、物理形态及药物和辅料的粒度。
制剂中辅料影响药物稳定性的常见途径为改变制剂中的水分、改变制剂中微环境的pH值、充当了广义酸碱催化剂、直接与药物发生反应或者成为了杂质来源,而这些杂质能够直接与药物反应或在药物降解中充当了催化剂。辅料还会通过离于交换作用、多晶型的转化、低共熔物或固态溶液的形成等方式改变药物的物理化学形态。这些物理或化学状态的改变都会影响药物的化学稳定性。
在固体相容性试验中,辅料的两个性质对于处方稳定性和相容性试验是尤为重要的,即为:
①在不同湿度环境下的吸水能力;
②引入辅料后的pH值。
2.1水分和微环境pH值的影响
大多数的药物和辅料都含有水分,这些水分以结合水或非结合水的形态存在,结合水是指紧密结合到物质物理形态上的结晶水,它几乎是不能移动并且不可反应的。例如,B-内酰胺类抗生素水解不稳定,其结晶水合物却是稳定的,原因在于这些水是结合在晶体基质中且不可反应的。正如预期的那样,这类化台物的稳定性高度依赖于它们的结晶状态”。相反,非结合水通常存在于一种平衡状态中,并且有较高的分子运动性。辅料中水分随湿度的变化即反映了非结合水的含量。
水分在辅料或药物和辅料混合物中的物理状态决定了其在药物与辅料相互影响中的潜在作用。辅料对水分的吸附解吸性质的影响已得到充分证实。固态系统中的水分会对稳定性有重要影响,这些影响不仅体现在可能引起药物(如乙酰水杨酸)的水解;还在于水分的参与使其作为反应介质,增加了系统的塑性和分子流动性。吸水能力强的辅料可以通过在封闭系统中清除水分的方式,预防药物降解。与一些低吸附能量的辅料相比,具有较高吸附能量特性的辅料能够降低系统中水的反应活性。而另一方面,有些辅料(如微晶纤维素)由于其弱吸附性,其中的水分又是具有高度反应活性的。原因在于:相对超细纤维来说,微晶纤维素会引起阿司匹林水解速率的增大。
基于辅料带来的固体表面微环境pH值对药物化学稳定性有重要影响。由于辅料自身的化学性质和组成,它们有酸性或碱性的表面pH值。对于可溶性辅料,辅料溶液的pH值对其在固态时pH值的简单指示。而对于不溶性辅料,其中5%~20%的辅料混悬状态的pH值可以作为间接指示。基于处方前溶解性和稳定性研究,选择具有合适pH值的辅料,对辅料相容性实验设计非常有帮助。例如,呈碱性的硬脂酸镁可能导致碱敏感药物不稳定。
大多数药物是有机酸/碱形成的盐,它们在酸性或碱性pH值条件下以游离酸或碱存在。少量药物会溶解在自由水中,故pH调节剂会导致药物游离酸碱基的形成。如果游离酸碱基不如其成盐形式稳定,则些时会引起药物加速降解。当然这也有可能因药物挥发或升华,以药物损失的方式引起制剂失重,而非降解产物的存在。
2.2辅料及其杂质的反应
一般来说,药用辅料被认为是惰性的。但其也是具有功能基团的有机化合物,在制剂中有可能发生化学反应,尤其是与药物活性成分( APIs)的活性基团发生反应。而且,药用辅料所含微量活性杂质也会催化或直接参与药物降解反应。
对辅料合成、分离和/或纯化进行简要考察,将会得到一些重要提示,即其潜在杂质和可能带来制剂稳定性问题的其它特性。但因其专有性,这类信息难以获得,并大多受限于非正式的供应商讨论和对专利数据的精读。文献提供了一些药用辅料活性杂质的例子可作为指导(表1)。以下列出了其中几个实例。
表1 常用的药用辅料生产方法及潜在的活性杂质


①众所周知,还原糖(如乳糖)与伯胺和仲胺类药物会发生美拉德反应。接着通过阿马多尔重排生成一系列有色产物。这些反应的机理为:胺类化合物与开链形式的碳水化舍物生成一类亚胺离子中间产物,进而可闭合生成葡糖胺或去质子化生成烯醇形式的重排产物【图1(a)】。
图1 药物与药用辅料及其活性杂质的反应实例
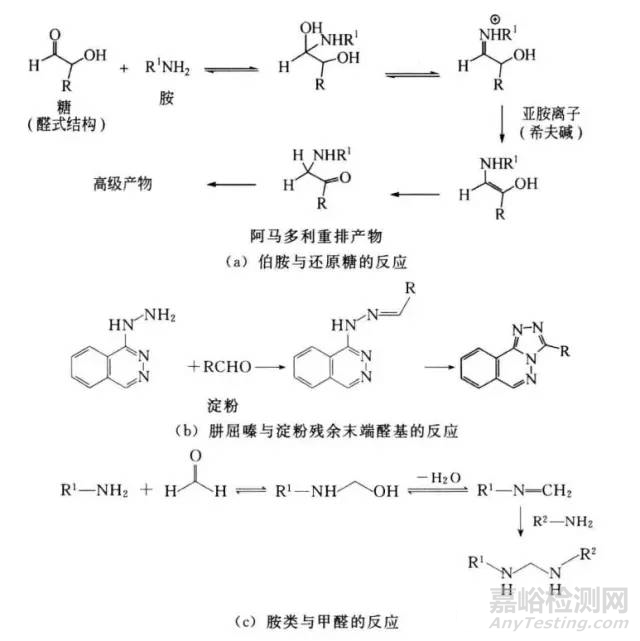
②即便是非还原糖,也可能含有微量还原糖。据报道,淀粉这类终端葡萄糖也会与制剂中肼屈嗪发生反应【图1(b)】。
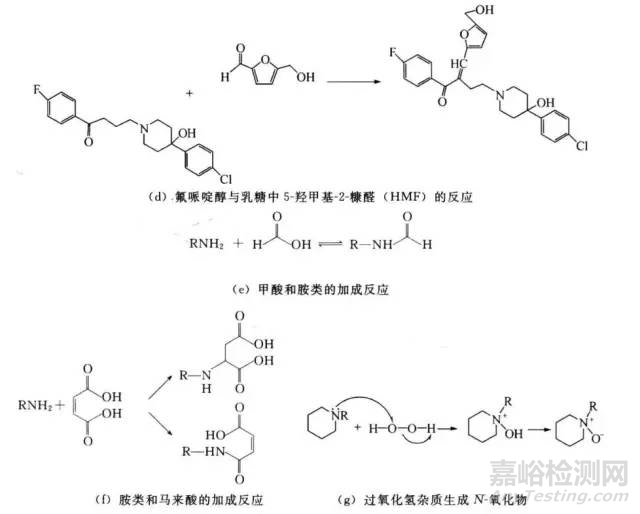
③一些辅料和包装材料中,存在甲醛和其它醛类物质等杂质。甲醛会和胺类药物反应生成N-甲酰产物,进而生成二聚物【图1(c)】。阿德福韦会与甲醛反应生成活性亚胺,亚胺会与另一个氨基分子发生亲核加成反应生成二聚物。Nassar等人指出,BMS-204352与助溶剂(吐温80和PEG 300)中的甲醛杂质生成加合物。据报道,乳糖中的一种杂质——5-羟甲基-2-糠醛会与氟哌啶醇反应生成缩合产物【图1(d)】。
④甲酰物可与胺类反应生成甲酰胺【图1(e)】。例如,Waterman等人报道瓦伦尼克林。一种仲胺化合物,会在渗透泵片剂中聚合物的活性杂质(甲酰和乙酰物)作用下发生N-甲基化和N-甲酰化反应。而氟哌酸硬脂酰衍生物的生成则与硬脂酸镁的硬脂酸盐成分相关。与图l(e)所示机制相似,氟哌酸的仲胺会发生亲和加成反应,生成硬脂酰胺。塞罗西汀会与其平衡离子发生迈克尔( Michael)加成反应,生成两种加成产物【图1(f)】。
⑤在含硬脂酸镁的片剂中,福辛普利钠的降解是由镁离子螯合作用造成的。
⑥具有醇基的药物能与酸(如甲酸)生成酯,或者可与酯类(如对羟苯甲酸酯/尼泊金)发生转酯(基)作用。同样地,酸性药物可与含醇基的辅料(如PEG类)发生酯化反应。
⑦制剂中即便有痕量级的过氧化物和金属离子,也会加速药物的氧化作用。残留过氧化物存在于聚乙烯吡咯烷酮(PVP)、交联PVP、羟丙基纤维素(HPC)中。此外,它们的含量随不同批次和厂商而变化。过氧化物能引起自由基启动的氧化反应,能与叔胺发生亲核加成反应生成N-氧化物,与伯胺反应生成羟胺,与硫化物反应生成硫氧化物/亚砜。例如,PVP中过氧化氢杂质与哌嗪环反应生成了N氧化物【图l(g)】。
⑧羟乙酸钠是生产超级崩解剂——羧甲基淀粉钠过程中残留的一种反应物,能引起度洛西汀等药物的降解。
⑨二氧化硅含显著水平的重金属杂质,会在一些氧化降解反应中起催化剂作用。
2.3稳定剂
尽管相容性研究目标在于药物辅料相互作用引起的潜在或严重的稳定性降低等方面,但辅料也经常被用于提高制剂稳定性。例如,一些实例证实了环糊精包合物可以改善药物的不稳定性。它能将不稳定药物包台到疏水空洞内部,形成保护层使其免受水解、氧化和光降解等降解机制的影响。
通过在制剂中加入抗氧化剂,是一种众所周知的使氧化敏感药物稳定的方法。选择的抗氧化剂最好是水溶性(如没食子酸丙酯和维生素C)或水不溶性物质[叔丁基对甲氧酚(BHA)、二叔丁基对甲氧酚(BHT)或a-维生素E]。而且抗氧化剂的选择不仅基于它们的性质,而且也要基于其氧化机制。因此,相容性研究通常包括不同氧化剂改善药物降解的相对效果研究。
此外,制剂中重金属催化降解反应通常可通过鳌合剂的使用而得到改善,如乙二胺四乙酸(EDTA)或乙二醇四乙酸( EGTA)。相比EDTA,EGTA与钙、镁离子的亲和力更强。例如,右美沙芬与离子交换树脂、二乙烯基苯磺酸的混合物中。EDTA的加入可改善其氧化降解反应。
直观而言,要使水解敏感药物稳定,则不允许选择有高残留水分和高吸水能力的辅料。但是,水亲和性的辅料也有可能优先吸收透过包装的水分,从而改善药物在保质期和加速储存期的水分敏感性。例如.在固体制剂中使用硅胶以提高对水极敏感的克拉维酸钾的稳定性。
光照能通过不同机制导致药物降解,如不饱和体系的加氢反应、聚合、同分异构化、光氧化和取代反应。耐光材料的使用,如棕色玻璃和不透明的高密度乙烯( HDPE)瓶,是光敏感药物的标准保护方法。此外,在药品生产和包装过程中,耐光薄膜包衣和环糊精、染料及有色添加剂等辅料的使用通常也是有效的。而光敏感辅料也可用于光敏感药物制剂中,正如易氧化药物中的抗氧化剂作用一样。在一些实例中还发现,和药物的紫外吸收光谱重叠的辅料有助于提高药物稳定性,如维生素B2或姜黄素能提高硝苯地平的稳定性,羟甲氧苯酮能提高磺胺异二甲嘧啶的稳定性。
3.实验设计
相容性研究涉及一系列设计方案,旨在确定关键药物一辅料的不相容性及其原因(图1)。关于新分子实体的相容性研究,总是始于对现有资料和候选药物化学结构的评价,用以确定分子“薄弱点”。活性或不稳定官能团的存在、pKa值和已知活性的相似化合物都为辅料的选择提供了有用的信息。除一股文献外,现在有若下个计算程序可用于帮助预测候选药物的潜在降解途径,如CAME、SPARTAN、EPWIN0和PhARM。许多制药企业也有内部的数据库和软件程序。而且.关于物理化学性质和药物分子强制降解的处方前研究,也被用于调整相容性研究的设计以检测已知反应发生与否及其进行程度。
相容性研究设计可能涉及药物与一种或多种辅料的混合。将这些混合物以本身物理混合物或压制后的形式,在不同的加速条件下培养。在这些究体系中,常加人水以评价它在加速药物与辅料之间相互影响中所起的作用。而其它成分的添加是基于分子敏感性的背景资料。如加入双氧水以诱导氧化应激。相容性研究的样品通常在高温条件下储存,并在预设时间间隔内分析药物的物理和化学变化。此外.采用热处理方法分析药物和辅料的二元混合物,如用差示扫描量热法(DSC)和等温微量热法(IMC)快速评估它们潜在的不相容性。简而言之.相容性研究涉及试验每个阶段的若干个选择,它们取决于候选药物、可用文献和研究目的。以下部分将重点介绍这些决策的基础。
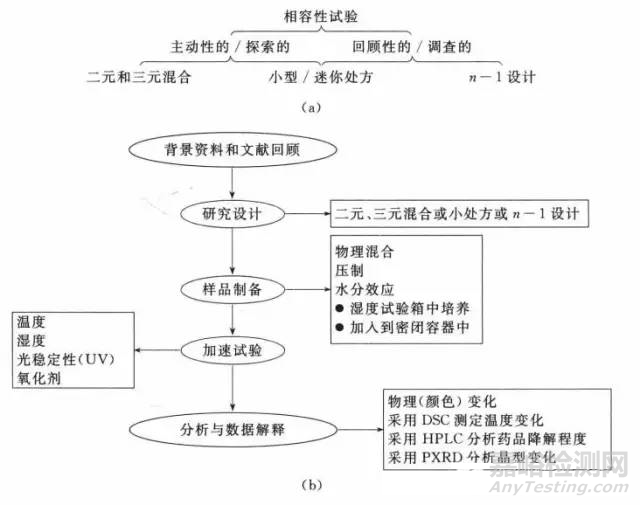
图1 相容性试验(a)和研究实施(b)的典型模式(椭圆形表示相容性试验的不同阶段,方框表示每个阶段的关健因素和变量)
相容性研究通常通过加速试验来开展,且对其作用的评价是基于药物辅料的二元或多元混合物。实验设计由可能选择的处方和首选的辅料来决定。这些决策与所有其他可用的处方前数据、药物活性成分(API)的特性及市场偏好相关。这些也决定了评估多种药用辅料的类型。例如,不溶性化合物液体制剂处方的相容性研究包括了表面活性剂和混悬剂等辅料,这与水溶性好的化合物的研究设计大一相同。
3.1二元或多元体系
通常,主动性的处方前相容性研究主要以二元或三元体系进行开展。将药物与辅料组成二元混合物(如药物与填充剂等常用药用辅料)或三元混合物(如药物、稀释剂和较低比例的崩解剂或润滑剂等辅料),长时间置于温度和湿度加速的条件下,并采用单独的药物和单独的辅料作对照。此外,由药物分子的特性决定的光照和过氧化物等恶劣的条件也包含在研究设计中。通过以下两个方面进行不相容性的评价:目测颜色或物理形态.以及光谱和量热方法来确定物理变化;对药物和杂质含量进行分析来测定化学变化。
3.2 n-1设计和小型/迷你处方
通常相容性研究目的是为解决处方的稳定性。该研究不包括每小批中只有个组分的情况以确定不相容性的来源。一般小型处方的制备不包括非关键性、小量和/或易相互变换的成分,如颜色、口感、从溶液到混悬液的情况。液体处方开发的相容性研究常常是小型处方研究,原因在于:在相容性试验和基本处方中使用适宜的缓冲系统之前,需要对其pH溶解性和稳定性进行评价。
普基特缅设计(Plackett-Burman design)可以用于包含小型处方的设计研究,尽管实际上它很少被应用。这项设计减少了实验进行的次数,可以找到引起主要不相容性的辅料。它可以在n+1次实验中检测n种辅料。迪里希和法西赫使用这项设计来考察盐酸吡哆醛与11种辅料的相容性,在两种温度(25℃和55℃)和两种湿度条件(11%和75%RH)下,只进行了16次实验。这项研究包含8次实验,超过了研究“伪变量”影响的最低要求,以解释随机实验误差。然而,这种方法未考虑到根据混合物存在的组分个数而变化的辅料浓度。
4.样品的制备
4.1 样品的制备和储存
相容性研究中样品的制备取决于各成分的物理性质和最终处方的概念化。对于二元相容性研究而言,选择合适的药物与辅料的比例常常基于最终处方中预计使用的重量或物质的量。在缺乏药物确定剂量时,通常会是在新化合物分子的早期开发阶段,应该测试最糟糕的情况,即最小的药物辅料比。
使用最小的药物/辅料比,以其能提供药物与辅料最可能发生的相互作用。例如,默克公司的化合物L-649923无定形碳酸钙盐,是一种白三烯D4拮抗剂,可通过分子酯化降解形成R-内酯。当用微晶纤维素和预糊化淀粉作辅料时,药物剂量较低时其降解率很高。这些结果符合之前的假设,即反应发生在药物与辅料的界面处。较小粒径的辅料导致药物降解率较高,也进一步支持了该假设。因此,含二水磷酸氢钙的片剂中,乙酰水杨酸的水解也表明了与辅料粒径的秩相关性。同样,粉末型硬脂酸镁对药物降解水平的影响高于颗粒型。
4.2 样品的制备
固态样品的二元混合设计通常只涉及物理混合。必须重视药物和辅料细颗粒的应用;如若需要,也要注意每个组分的松团作用。通常,通过共同过筛来达到各组分的均匀有效混合。将药物与辅料的混合物压制后,进行固态相容性试验。但晶体的研磨和紧压可以形成无定形态。工艺应力如压制、研磨和干燥也可导致活性成分和辅料失去结合水。例如,研磨可以除去茶碱水合物中的结晶水,还会影响氨比西林和硫酸普拉睾酮钠的固态稳定性。同样,经球磨机研磨后,头孢克肟的结晶度降低并发生脱水。但是,对于湿敏感、低熔点的化合物以及需要通过研磨来降低粒径的药物而言,相容性研究所包含的研磨和压制是有益的。
水分的存在往往加速降解动力学。此外,吸附的水分可加快异构化和/或结晶过程。处方前相容性试验的重要目标之一是确定工艺条件的可行性,如温法造粒和包装的防潮需要。这样,确定辅料存在时药物的湿敏感度就变得很重要。为此,在封闭体系中,常将水加入药物与辅料二元混台物以创高相对湿度(RH)环境。或者在不同温度和RH条件下,将这些混合物储存在敞口容器中。在比较这些设计试验中的数据时必须谨慎,因为储藏在敞口容器的条件下,潜在的挥发和活性成分的损失可能给出的是稳定迹象,但在最后剂型中也许并非如此。
对于相容性试验所采用的加速环境储存条件的选择,是基于API的特性与最终制剂在储存期间所预期的应力。这些包括储存在高温、高湿条件下,暴露在紫外光照和过氧化物中等。虽然国际协调委员会(ICH)和管理机构提供了终剂型加速储存条件选择的指南,这些指南是针对基于目标标签储存要求而确定的稳定性研究,但相容性研究加速条件的选择仍由处方设计人员决定。
4.3 热应力
对相容性研究而言,恒温压力测试(IST)或样品在较高的恒温下培养(作为加速条件)几乎是很普遍的。这是基于以下假设,即:降解反应动力学遵循阿伦尼乌斯动力学。
选择较高的温度可以显著加快反应率,因此即便是相对缓慢的反应在很短时间内也变得明显。例如,假设固态反应的平均活化能为105kj/mol,那么在25℃放置约5年的药物降解量相当于在60℃放置3周。
通常,通过升高温度来增大反应速率,从而缩短试验时间。然而,在选择温度时必须要小心谨慎,因为超过一定温度,系统将超出替代降解途径的活化能。这样会得到非代表性的数据。HPLC图谱上额外杂质峰的出现常常表示主要降解过程的发生。例如,在探索实验化合物的加速降解条件时,Sims等观察到当培养温度上升到80℃时,得到与在室温和60℃条件下相同的杂质,样品在100℃时则出现许多不相关的峰。因此,通常选择多个刺激条件,以确定在高温下观察到的降解反应是否有这么高的活化能。而只有在与药物无关的情况下才发生。
4.4 湿度和/或水分含量
相容性研究的高水分含量不仅为了揭示水直接参与的反应(如水解),而且还旨在研究水是否会提高固体反应活性。作为反应物的介质或增塑剂,固体表面的吸附水可提高反应活性。固体组分混合物中水的存在,为两个固体成分的反应提供了所需的分子流动性。
可通过以下方式来考察水分对辅科相容性研究中样品的影响:
①制备混悬液;
②在个封闭的体系中加水(通常为固体量的20%);
③将体系暴露在湿度可控的条件下。
这些方法各有利弊。混悬实验可以为模拟混悬剂和快速评价药物底物的水敏感性提供有用信息,它们通常不模拟固体制剂条件——这对回顾性或研究性的兼容性研究的数据解释是很重要的。封闭体系中,在药物与辅料的混台物中加入固定比例的水,决定了体系中水的初始含量.并能部分模拟湿法制粒条件。然而,样品中含水量的变化取决于以下两个因素,即储存温度下水分的吸收动力学和样品平衡水分的含量。而且空气中相同的含水量致使高温时相对湿度较低。相比而言,可控湿度条件下样品的培养保证了固态体系中平衡水分含量的相对恒定,这样使水作为介质或只是增加反应物的分子流动性的降解反应研究得以进行。然而,敞口容器研究可能导致反应中的活性挥发性杂质从混合物中耗损,这样可能改变反应途径。此外,在恒湿试验箱中(如敞口碟)储存样品,可能导致诱导期的增加,从而使该实验中的动力学比加水实验(或混悬试验)中的更为复杂化。
鉴于每种试验方法的利与弊,制剂研究人员必须要在现有处方前数据的基础上,为每项相容性研究设计选择合适的条件。
4.5 机械应力
机械应力往往是药品生产工艺条件中不可避免的部分,如研磨和压制。这些力可能导致药物无定形孔笼或药物分子晶体晶格缺陷的形成,或简单地增加药物-辅料混合过程中的亲和性和接触面。因此,机械应力会导致降解反应率的增加。例如,在相容性研究中,将普鲁卡因青霉素G与无水磷酸氧钙和微晶纤维素研磨,其降解率几乎为未研磨的药物一辅料混合物的两倍。同样,巴达维等人发现,相比于未压实的二元混合物,在研的化舍物DMP-754与无水乳糖压实后水解率提高了3倍多。
除化学反应外,压制对药物的物理形态也有影响。郭等人发现,盐酸喹那普利在压制后,其结晶度有所损失。由于其在乙腈(ACN)中形成晶体,故晶格中松散地掺有ACN,经压制后随着AcN的损失,导致结晶度降低和环化降解物增加。该现象的原因在于:相比结晶态,形成的无定形物发生环化反应所需活化能显著降低。
当怀疑在研药物可能会不稳定时,需要对其进行机械应力作用的研究,如亚稳的多晶型或已知的该分子化学反应情况。在相容性研究中,机械应力的模拟包括使用球磨机研磨、药物辅料混台物共研磨等操作。此外,还通常使用卡弗液压机来模拟压片和干法制粒的压制过程。
4.6 氧化应激
在药物体系中,氧化是药物降解的最常见原因之一。通常,氧化降解表现出药物降解的独特模式,例如,诱导期后杂质的快速增加,对微量自由基的敏感度。如果处方前或API特性研究显示其具氧化敏感性,可设计相容性研究以综合评价氧化敏感的程度,并找到解决的办法。这些方法包括如下一种或多种:
①在药物辅料混合物中加人不同浓度的氧化剂,如过氧化氢、金属杂质(铜和铁盐)或不同浓度的自由基引发剂;
②将相容性实验样品置于密闭容器的空气、氧气、氯气或氩气中,对药物降解进行比较研究;
③加入自由基清除剂和重金属螯合剂;
④包装配置,包括加入氧清除剂和具有最低渗透率的材料;
⑤使用不间批次已知残留过氧化氢含量的辅料,如PVP、交联PVP、HPC、吐温80和PEG-400;
⑥不同抗氧化剂的研究,如丁基羟基苯甲醚(BHA)、丁基羟基甲苯(BHT)、a-生育酚、没食子酸和抗坏血酸。
这些研究样品的制备中,如压制,应小心控制制备条件。而且,辅料中的过氧化氢杂质通常显示批次间的差异,使得批量试验中杂质含量或高或低。通常需开展叠加试验,以量化辅料中过氧化氢杂质的容许限度,这可以用于设定进料规格。
5.样品分析与数据整理
一套设定的相容性研究的试验与主要结果检测不仅取决于所设想的最终剂型及产品形式,也取决于候选药物在化学和处方前研究方面的可用背景资料。大多相容性研究包括任何颜色变化的外观检查、压制物/片剂的完整度与潮解作用、监测药物降解的定量化学分析。此外,还应检测活性成分在候选辅料中的晶型变化。
5.1 检测药物的降解
加速条件下对相容性样品进行目测,应观察包括颜色、气味、潮解、粉末流动性等方面的改变,这些观察的内在主观性通过对照组和冷藏组的秩相关来克服。可以采用紫外-可见光谱进一步量化颜色的变化,但是,这种方法也有这样的局限性吸光度的微小变化不会表现出显著的差异,即便在降解存在的情况下,降解物可能保留药物的生色团,从而导致吸光度的微小或无变化。尽管如此,当观察到显著差异时,这些观察结果是组分间发生不相容性的很好的说明。
几个药的外观产生了变色,表明其不稳定性和/或不相容性,包括如异丙嗪、去氧肾上腺素、克拉维酸钾、头孢呋辛酯和特比萘芬。常见的例子是处方中含有伯胺或仲胺和还原糖,如乳糖时,处方会变色,往往变为棕色。这通常伴随有美拉德反应,其终产物经历Amadori重排后形成具有颜色的中间体和终产物。
HPLC的紫外检测器是目前量化药物降解的最常用方法,它可以测定药物的含量,总杂质或储存条件下随时问推移特定杂质的增长。测定单个或所有杂质的增多往往优于测定药物含量的减少。因为大量的微小改变降低了数据处理的敏感性,如药物作用强度从99.5%降低到99.2%可能被认为不显著,但杂质含量从0.02%增加到0.32%则认为是显著性增加。
5.2 热分析法
诸如HPLC等定量、稳定性指示分析方法并不适用于药物开发初期阶段。在这种情况下,热分析法不失为快速、非特异性、劳动密集低的筛选工具。但要注意的是,从热分析法中获得的结果通常不是结论性的,必须经其它独立观察来难证。
相容性试验的热分析法依靠样品的吸热或放热的能量变化。相容性研究中最常应用的热分析技术是示差扫描量热分析法(DSC)。它包括在控制的方法下加热或冷却样品,由样品的温度来测定其热量的释放或吸收,参考标准同样随时间变化。另一方面,可采用等温微量热法(IMC)测定样品池中的热液并与参比池中热流相比较,因为两者都维持在恒温。IMC可以用于检测非常缓慢的反应,这样药物-辅料混合物可在室温或低温下检测,它也可以检测“实际生活”中重要反应。
相容性评价的DSC分析包括记录在标准加热速率下单个辅料、药物及其物理混合物的热相图,所有操作通常在氯气中进行。这些曲线简单的叠加可以解释并确定混合物的热性能是否为各组分的总和,或认为是非相互作用组分的基本属性。通过转变温度,峰形,峰面积的变化确定转变峰的出现和消失变化从而鉴别相互间作用。但是,应谨慎解释DSC的结果以确定辅料相容性,应结合应用其它技术如红外(IR)光谱和恒温应力测试(IST)。
Verma和Garg展示了一个有趣的研究实例,即物理观察,IR光谱和IST与DSC联合应用确定辅料相容性。他们记录了药物格列吡嗪和辅料微晶纤维索(MCC),乳糖和Tris缓冲液在1:5,1:5和药物、辅料各自1:1时的DSC热相图(图2),在这项研究中:
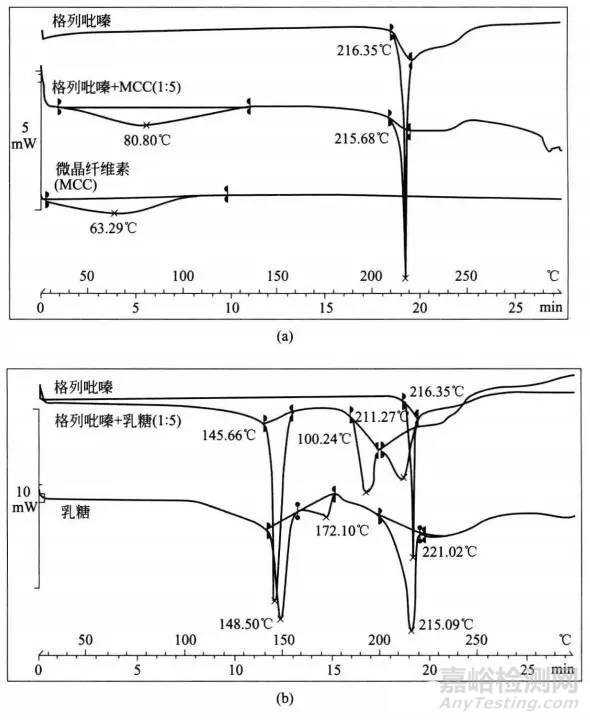
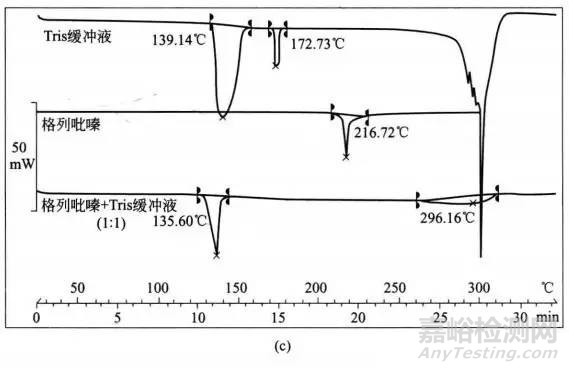
图2 采用DSC进行辅料相容性评价的应用实例(a)格列吡嗪,MCC和格列吡嗪、MCC混合物(1:5)的热分析图;(b)格列呲嗪,乳糖和格列吡嗪、乳糖混合物(1:5)的热分析图;(c)格列吡嗪,Tris和格列吡嗪、Tris混合物(1:1)的热分析图。
①MCC的DSC热分析图表明由于吸附水的损失,在63.29-时存在宽吸热,格列吡嗪在216.35℃时表现出熔融的吸热峰。两种辅料的物理混合物都出现了格列吡嗪和MCC的吸热峰,表明它们之间无相互作用。物理混合物的IR光谱显示了格列吡嗪的特征谱带,未见新的谱带,证实格列吡嗪与MCC是相容的。
②乳糖的热分析图在148.50℃(吸附水脱水)、172.10℃(晶型转变)、215.09℃,在221.02℃时出现一个小峰。物理混合物的热分析图未出现药物熔融峰,表明它们之间发生相互作用。混合加热后变黄色进步证实了它们的不相容性。
③Tris的热分析图在139.14℃(失去吸附水)和172.73℃(熔点)出现两个吸热峰。300℃时波浪式吸热表明Tris缓冲液的分解。在物理混合中Tris缓冲液的峰转移到较低温度(135.60℃),药物峰消失,在296.16℃时观察到一个宽的驼峰。尽管消失的药物峰表明药物-辅料之间相互作用,但是物理混合物的IR光谱仍然显示出格列吡嗪的特征谱带,未观察到新峰的产生,表明没有发生化学作用。
当药物-乳糖的特理混合中未出现药物峰时表明其不相容性,因为这可以由颜色变化的物理观察中证实;在药物-Tris缓冲液物理混合物中,无药物峰不能解释其不相容性,因为缺少了IR光谱和其它方法的相关依据验证。
5.3 晶型变化的检测
药物的发现与早期阶段的开发工作不仅是为了选择正确的药物分子,而且要确定其最佳剂型。药物的物理形式、多晶型及其盐型对候选药物的物理、化学、机制和生物药剂学特性等方面都有重要的影响。例如,较高的热力学活度状态,即无定形一般具有较高的表观溶解度、扩散溶出速率及较强的吸湿性和化学反应度,同时它也有向更稳定晶型转变的趋势。有关相变的机制已在文献中报道。
在药物开发过程中非常重要的一点就是要确保制剂组成及工艺条件不会改变或水破坏药物的物理化学形式。必须注意的是,尽管这些研究不是兼容性试验的常规部分,但如果开发晶体的亚稳形式或关于API稳定性的资料可以监测剂型变化时,这些研究就变得非常重要。
6.结论
辅料相容性研究的主要目的是选择与药物相容的剂型组成。有条不紊地进行实验并提供药物稳定性方面的其它信息,并确定降解产物及其机制。而且,如果发现药物的稳定性不好,应采用一些策略以降低药物的不稳定性。本文提出的方针和原则将有利于在相容性研究中进行合理的实验设计、实施和解释,以加速处方的开发并防止或尽量减少药物开发中的风险。

来源:研药技术交流汇


