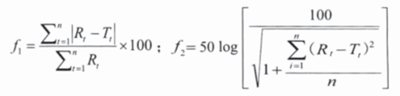溶出度试验技术是评价口服固体制剂内在质量的一种重要手段,该试验不仅可为建立体内外相关性提供基础数据,而且有望成为通过体外试验评价口服固体制剂质量的简易、有效可行的方法。
在多种 pH 溶出介质中溶出曲线的测定是先进国家药物审评机构评价口服固体制剂内在质量的一种重要手段。
该试验可用于评估不同来源的同一制剂内在质量差异。日本自 1998 年开始实施“药品品质再评价工程”,陆续出版了《医療用医薬品品質情報集》( 即日本参比制剂目录、橙皮书、Orange Book),其中详细罗列了所收载制剂的四条标准溶出曲线。
美国 FDA 药品审评中心的仿制药办公室属下的生物等效部也于2004年1月起,在其官方网站上推出了“固体制剂溶出曲线数据库”。
目前我国虽未推出“溶出曲线数据库”,但对口服固体制剂多条溶出曲线的测定已愈发受到关注与重视。
本文在参照国际药物联合会 (FIP) 颁布的《口服固体制剂溶出度试验指导原则》、日本厚生劳动省颁布的《仿制药生物等效性试验指导原则》、FDA 颁布的《常规口服固体制剂溶出度试验工业指南》、美国药典 31 版《溶出度试验验证法:研究与建立》( 附录 1092) 和欧洲药典 2008 年版《溶出度试验法》( 附录溶出度试验法 2.9.3) 基础上,详细阐述了如何采用多条溶出曲线评价固体制剂内在质量的具体试验步骤,希望能通过此种检测手段找到国产固体制剂与进口原研制剂的差距,为临床疗效的差异提供佐证,为国家相关技术法规的拟定与完善、为企业切实有效地提高药品质量提供依据。
一、采用多条溶出曲线剖析参比制剂
原研制剂的确定
由于原研制剂 ( 亦称参比制剂 ) 一般皆为国际制药公司开发研制,在质量水平及其控制上有严格的要求。我国绝大多数药物制剂均为仿制药,只有质量和原研制剂相同或相似,才能保证其用药的有效性和安全性。
其中溶出曲线的对比,也应以原研制剂作为“参比”。所以,试验前一定要获得原研制剂,以其作为参比制剂进行测定。
对于参比制剂的遴选,原则上应从多批号样品中撷取一“标准批号”,由于客观条件的限制,建议最低要求应获取一个在有效期内批号的样品进行研究。
溶出介质的选择
1 普通制剂
酸性药物制剂:pH 分别为 1.0 或 1.2、5.5~6.5、 6.8~7.5 和水。
中性或碱性药物 / 包衣制剂:pH 分别为 1.0 或 1.2、3.0~5.0、6.8 和水。
难溶性药物制剂:pH 分别为 1.0 或 1.2、4.0~4.5、6.8 和水。
肠溶制剂:pH 分别为 1.0 或 1.2、6.0、6.8 和 水。
以上根据药物酸碱性而设定的相应 pH,参照了日本《仿制药生物等效性试验指导原则》中的溶出度研究内容。其中对难溶性药物制剂则是根据美国 FDA 公布的《仿制药指导原则》,但本文将 4.0 扩展到了 4.5。
2 缓 ( 控 ) 释制剂
pH 分别为 1.0 或 1.2、3.0~5.0、6.8~7.5 和 水。
3 讨论与说明
药物 pKa 小于 3.0 的可看作酸性药物,大于或等于 3.0 的可看作中 / 碱性药物。
在进行溶出曲线测定之前,应首先测定主成分在各种溶出介质中的溶解度,以确定该制剂是否为 “pH 依赖性制剂”。日本橙皮书中收载有这些数据。
试验前还应进行原料药在不同 pH 溶出介质中的稳定性考察,以确保试验数据的准确测定。日本橙皮书中收载有该部分内容。
以上含有 pH 范围的,可分别按 0.5 或 1.0 间隔测试。如溶出曲线差异较大,应考虑分别试验;如无明显差异,酌情选择即可。
美国一般采用 1.0、4.5、6.8 和水,日本一般采用 1.2、4.0、6.8 和水。无论何种制剂都不宜采用 pH 8.0 以上的介质进行试验。如确有必要,应提供充足理由。
FDA 公布的溶出度数据库中,只有阿维 A(acitretin) 胶囊采用 pH 9.6 溶出介质。
但对于含有明胶交联的制剂,由于此类制剂的生物利用度与交联度呈正相关,在溶出介质中加入适量酶,有助于评价该制剂的溶出度。建议研究时可考虑添加酶以及酶的种类与浓度。
溶出介质的配制
1 欧洲药典
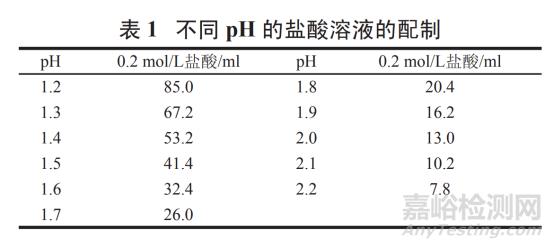
pH 1.0~2.2 盐酸溶液:
精密量取浓盐酸 9.0 ml,加水稀释至 1 000 ml,摇匀,即得 pH 1.0 的盐酸溶液。其他 pH 盐酸溶液:按表 1 量取 0.2 mol/L 盐酸 ( 精密量取浓盐酸 18.0 ml,加水稀释至 1 000 ml,摇匀,即得 ) 适量,加水稀释至 200 ml,摇匀,即得 pH 1.2~2.2 盐酸溶液。
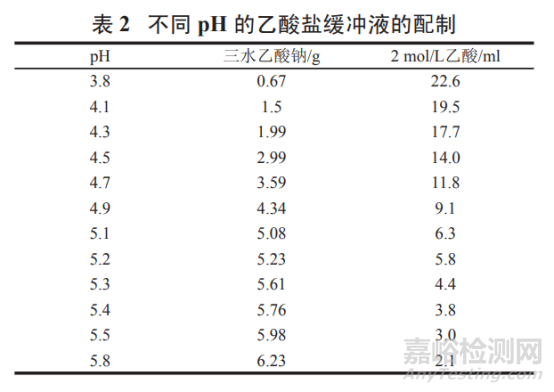
pH 3.8~5.8 乙酸盐缓冲液:
按表 2 取三水乙酸钠和 2 mol/L 乙酸 ( 取冰乙酸 120.0 g,加水稀释 至 1 000 ml,摇匀,即得 ) 适量,加水溶解并稀释至 1 000 ml,摇匀,即得。
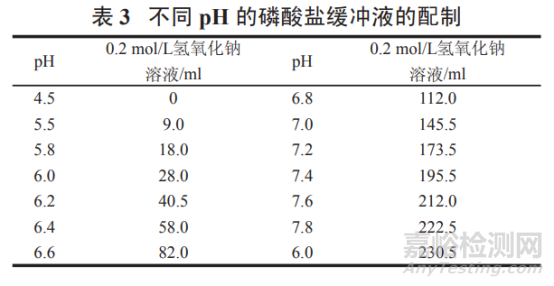
pH 4.5~8.0 磷酸盐缓冲液:
精密称取磷酸二氢钾 6.80 g,按表 3 加入 0.2 mol/L 氢氧化钠溶液 ( 取 8.0 g 氢氧化钠,加水溶解并稀释至 1000ml,即得 ) 适量,加水溶解并稀释至 1 000 ml,摇匀,即得。
2 美国药典
pH 1.0~2.2 盐酸 / 氯化钾溶液:
除需加入氯化钾 0.75 g 外,其他配制方法同欧洲药典。
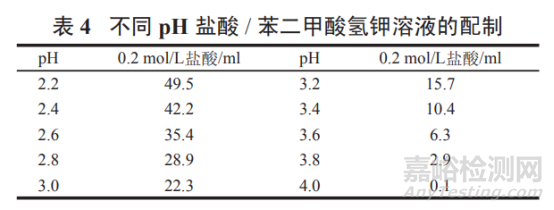
pH 2.2~4.0 盐酸 / 苯二甲酸氢钾溶液:
取苯二甲酸氢钾 2.04 g,按表 4 加入 0.2 mol/L 盐酸适量, 加水溶解并稀释至 200 ml,摇匀,即得。
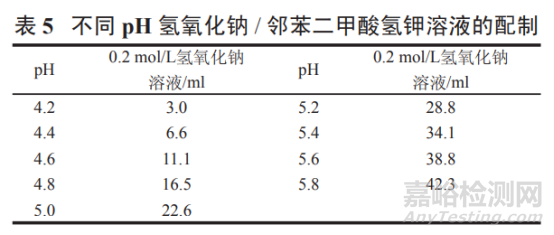
pH 4.2~5.8 氢氧化钠 / 邻苯二甲酸氢钾溶液:
取邻苯二甲酸氢钾 2.04 g,按表 5 加入 0.2 mol/L 氢氧化钠溶液适量,加水溶解并稀释至 200 ml,摇匀, 即得。
pH 5.8~8.0 氢氧化钠 / 磷酸二氢钾溶液:
同欧洲药典。
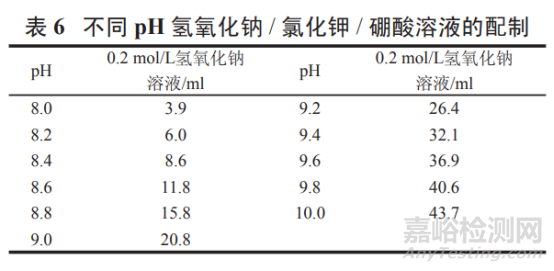
pH 8.0~10.0 氢氧化钠 / 氯化钾 / 硼酸溶液:
取氯化钾 0.75 g 与硼酸 0.62 g,按表 6 加入 0.2 mol/L 氢氧化钠溶液适量,加水溶解并稀释至 200 ml,摇匀,即得。
3 日本药典
pH 1.2 溶液:
取氯化钠 2.0 g,加水适量溶解后,加盐酸 7 ml,再加水稀释至 1 000 ml,混匀, 即得。
pH 4.0 溶液:
将 0.05 mol/L 乙酸溶液与 0.05 mol/L 乙酸钠溶液按 16.4 ∶ 3.6 比例混合,即得。
pH 3.0~6.0 介质 ( 除 4.0 外 ):
取十二水合磷酸氢二钠 17.91 g,加水溶解并稀释至 1 000 ml, 得 0.05 mol/L 磷酸氢二钠溶液。另取一水合枸橼酸 5.25 g,加水溶解并稀释至 1 000 ml,得 0.025 mol/L 枸橼酸溶液。用该枸橼酸溶液调节上述磷酸氢二钠溶液至所需 pH 即可。
pH 6.8 磷酸盐缓冲液:
取磷酸二氢钾 1.7 g 和无水磷酸氢二钠 1.775 g,加水溶解并定容至 1 000 ml, 即得。
4 中国药典
配制方法详见中国药典 2005 年版附录。
5 讨论与说明
建议根据原研制剂生产厂商的国别选取溶出介质配制法。各介质配制 pH 误差应控制在 ±0.05。
溶出曲线的测定
1 测定时间点和结束时间点的设定
对于测定时间点,普通制剂与肠溶制剂可分别为 5、10、15、20、30、45、60、90、120 min,此后每隔 1 h 测定。缓 / 控释制剂可为 15、30、45、 60、90 min 和 2、3、4、5、6、8、10、12、24 h。
对于结束时间点,在酸性介质 (pH 1.0~3.0) 中最长测定时间为 2 h;在其他各 pH 介质中普通制剂与肠溶制剂均为 6 h,缓 / 控释制剂为 24 h。
但当连续两点溶出率均达 90%(缓 / 控释制剂为 85% ),且差值小于 5%时,试验则可提前结束。
2 讨论与说明
测定时间点的拟定:
研究表明,相当一部分原研制剂的溶出曲线具有延迟、拐点等现象 ( 如辛伐他汀片原研制剂就有5~10 min的延迟释放 )。
提示:
在仿制研发与质量评价时应加以注意,故以上测定时间点拟定得较为“紧凑”。但当比较时间点确定后,对于仿制制剂,则可仅进行比较时间点的测定。
试验样品的生产规模:
由于口服固体制剂的生物利用度与生产规模密切相关,故一般情况下应不小于今后工业化生产规模的 1/10 或不小于 10 万个单位。
测定数量:
为达到统计学要求,规定每个品种的测定皆应选取 12 个样本。但鉴于目前溶出仪尚未有 12 个溶出杯装置,且考虑到工作量等诸多因素,一般测定 6 个样本即可。
溶出度试验参数的确定
1 装置与转速的确定
对片剂,建议采用桨板法、50 r/min;对胶囊剂,建议采用转篮法、50 r/min( 如采用桨板法,则建议采用沉降篮 )。
通常认为 50 r/min 与中老年人体内胃肠道蠕动强度基本一致。除非特指或特殊制剂,不宜采用更慢转速。一般采用大杯法,介质体积为 900~1 000 ml,不建议采用小杯法。
如溶出样品浓度过低,可采用加大进样量、同时加大流动相中有机相比例,使被测组分峰保留时间缩短、峰形尖锐,以提高检测灵敏度。
2 试验参数
在某溶出介质中,当结束时间点溶出量仍达不到普通制剂 90%、缓控释制剂 85%时,则可酌情放宽溶出度试验参数。建议先增加转速至 75 r/min,如未果,则可在溶出介质中添加表面活性剂,添加浓度应以 0.01% (w/v) 为起点、逐步增加,不建议采用 1.0%以上浓 度。
如仍未达标,则再增加转速至 100 r/min,但绝不允许添加有机溶剂。因为这将严重背离体内外相关性原则,同时大大降低区分效应。
添加表面活性剂时,应注意不同来源试剂可能会对试验结果带来显著性差异的情况,尤其是在使用十二烷基磺酸钠时,故建议在质量标准中注明试剂来源,以使试验重现。
溶出曲线的测定
1 测定法
采用紫外法测定时,由于存在着辅料、胶囊壳等诸多因素的干扰,尤其在短波长 ( 低于 230 nm) 处,建议采用两点相减法,即一波长为被测组分最大吸收波长,另一波长为远端无吸收波长。该法可大大降低辅料干扰,并在日本橙皮书中广泛使用。
当以上方法无法排除测定干扰,或吸光度低 于 0.25( 即使采用 5 cm 长距离测定池 ),建议采用 HPLC 法。
无论采用何法测定,皆建议对照品溶液浓度配制成 50%~60%释放量,以兼顾溶出液的不同浓度,尽可能缩小外标一点法测定误差。
2 样品处理
采用一个滤膜即可,可大大提高工作效率,最大程度排除滤膜吸附的干扰。
按规定,溶出液取出后应过滤,但考虑到滤膜吸附的影响 ( 特别是经微粉化处理的小规格难溶药物制剂 ),建议:
①如采用 HPLC 法测定,建议溶出液离心后,取上清液测定。甚至直接置液相小瓶中,静置后进样。这样,便可仅取溶出液 2 ml,该体积相对于整个溶出液 (900~1 000 ml) 体积的影响很小,故可省略其后的结果累积校正。
②如采用 UV 法测定,由于每一时间点均应至少弃去 5 ml 初滤液,故抽取体积应不少于 10 ml。由于体积较大, 则应进行结果累积校正,否则不利于准确判断。
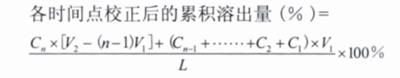
累积释放度校正计算公式
多次取样时,可采取及时补充同体积同温度溶出介质,亦可采取不补液两种方式,但必须保证每次抽取体积的固定性。累积校正计算公式如下:
1 补液
其中 Cn 为各时间点取出后的样品浓度 ( 即稀释前的 );L 为制剂标示量 (L/V2 单位需与 Cn 一致 );V1 为各时间点固定取样体积;V2 为溶出介质体积。
该公式如采用各时间点测得释放量表示,则可演变为:
其中 An 为各时间点测得溶出量。
2 不补液
计算时间点的确定
普通制剂如 15 min 内溶出量达 85%以上,则无需进行曲线比较。此时,仿制制剂亦应满足此条件。
只要 15 min 时溶出量未达 85%以上,需进行溶出曲线的比较。
溶出量在 85% ( 缓控释制剂 80%以上 ) 以上的时间点仅能选取一个;且由于所采用的相似因子比较法 —— f2 因子法计算结果有依赖于比较时间 点个数的特性,故建议选取溶出量间隔相近的 4~5 个 ( 普通制剂 ) 或 4~6 个 ( 缓控释制剂 ) 时间点进行计算比较,但这些时间点的间隔无需相等。
如上文所列试验,“当结束时间点溶出量仍达不到普通制剂 90%、缓控释制剂 85%时,建议先增加转速至 75 r/min,如未果,则可在溶出介质中添加表面活性剂,如仍未达标,则再增加转速至 100 r/min”。
若最终溶出量仍未达到标准,不建议再采用更为极端的条件。
此时,依据最终测定结果,选取溶出量间隔相近的 4 个时间点计算比较即可。
对于所选时间点溶出结果变异系数的规定
所选用的第一时间点溶出结果变异系数应不超过 20%,自第二时间点至最后时间点溶出结果变异系数均应不得过 10%。
若超出,应从仪器的适用性予以考虑解决,如增加转速或增加表面活性剂浓度,直至满足精密度要求。
二、测定仿制制剂
通过以上对参比制剂的测定,确立“溶出曲线测定时间点”后,在这些时间点分别测定仿制制剂。
仿制制剂的含量与参比制剂的差值应在 5%以内,且所选用的样品应在重量 / 装量差异所规定范围的 1/2 以内,以尽可能排除因个体差异给溶出度试验数据带来的影响。
其生产规模亦应同参比制剂。
如所选时间点的溶出结果变异系数超出规定,已说明该厂家产品批内差异的不良波动性。此时则无需再进行批间差异比较,但仍需计算出均值用于与参比制剂的比较。
三、溶出曲线比较法
目前,日本和美国官方皆规定采用相似因子比较法 ——f1 因子和 f2 因子法进行溶出曲线的比较。具体计算公式如下:
当用于不同来源制剂间比较时, ƒ2 因子应≥ 50;当用于同一来源制剂间比较时 ( 批间 / 批内差异、各种变更等 ),ƒ2 因子应≥ 65。
表 7 为溶出量平均差异与相应 ƒ2 因子临界值的关系。
由此可见,若直观评估、各时间点差异在 10%或 5%以内,则可基本断定 ƒ2 因子大于 50 或大于 65。
溶出曲线比较时,有时会出现仿制制剂溶出曲线明显高于 ( 或快于 ) 原研制剂,这并不能说明仿制制剂质量优于原研制剂,生物利用度试验才能最终判断两者是否具有等效性。
来源:《中国医药工业杂志》
作者:张启明 ,谢沐风,宁保明 ,庾莉菊