通过对国内外药品生产设备清洁验证的相关法规指南全面梳理,从质量管理体系及验证工作生命周期管理的角度对清洁验证进行了分析;明确了药品生产设备清洁验证要点并构建了要点结构图;对国内外药品检查中发现的药品生产设备清洁验证存在的缺陷进行了统计分析;对清洁验证常见问题及分布情况进行识别,为我国药品生产企业进一步做好药品生产设备清洁验证提供思路与参考,同时也为药品检查工作中对清洁验证针对性检查提供借鉴。
最大限度的降低生产过程中的污染和交叉污染是药品生产质量管理最为核心的内容之一。在药品生产过程中,普遍存在不同药品共用生产线和生产设备的情况,如何保证生产设备的清洁程序能有效去除残留物及污染物是避免污染与交叉污染的关键点与难点。而清洁验证是证明清洁程序能有效清洁设备并满足其预定用途的直接证据,也是确保避免污染与交叉污染,保证最终药品安全、有效与质量可控的关键内容。在药品生产的历史上,不乏由于清洁不彻底导致的药品质量事故[1]。在全球的药品检查中也发现药品生产企业关于生产设备清洁验证方面较容易出现缺陷,其中一些问题还可能影响药品质量,甚至导致药品安全问题。如何在质量管理体系及验证生命周期层面做好生产设备的清洁验证是保证药品质量的关键内容,有必要进行针对性的分析研究。
1、药品生产设备清洁验证法规指南概述及要点结构分析
1.1 药品生产设备清洁验证法规指南概述
清洁验证的目的是证明对生产设备的清洁工作能始终如一的将产品、溶剂及微生物残留清洁至可接受的限度,并阻止可能的污染与交叉污染[2]。针对清洁验证,国内外药品药品生产质量管理规范(GMP)均进行了规定,之间虽然存在一些差异,但总体原则与要求是一致的。世界卫生组织(WHO)、美国注射剂协会(PDA)、原料药委员会(APIC)、国际药品认证合作组织(PIC/S)等组织还针对清洁验证制定了对应的指导原则,如WHO Guidelines on validation-Appendix 3 Cleaning validation(2019) 、APIC Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants(2016)、PDA Points to Consider for Cleaning Validation(2012)、PIC/S Recommendations on Cleaning Validation(2007)。概括起来,法律法规及指南最核心的要求是对清洁方法进行验证,证实其清洁效果,其中应当充分评估设备使用情况、共线生产情况、清洁方法、最差条件、取样方法与取样回收率、取样位置、限度标准、残留物检验方法等因素,并开展必要的持续确认与再验证,同时需要关注标准清洁操作程序文件、清洁验证方案、日常监测等内容。
1.2 基于质量管理体系及验证生命周期的药品生产设备清洁验证要点结构
清洁验证是药品GMP中明确要求开展的工作,是确保对生产设备进行有效清洁的重要保证,其必然是药品生产质量管理体系的一项内容。清洁验证的开展必须遵循质量管理体系的各项要求,脱离了质量管理体系单独对清洁验证进行研究容易造成衔接性与体系性方面的问题。从清洁验证的全过程考虑,其本身就存在生命周期[3],包括清洁验证的设计与开发、清洁验证的实施、清洁验证的持续监测与再验证3 个阶段。科学、有效的生产设备清洁验证必须从质量管理体系及生命周期角度对关键要素进行管控,药品生产设备清洁验证要点结构图见图1。
图1 药品生产设备清洁验证要点结构图
清洁验证属于验证的一种,必须遵循验证的统一要求,应按照验证主计划规定及验证管理原则实施验证。在验证全过程遵循质量风险管理的要求[4],注意文件与记录的管理,并确保数据可靠性。对实施过程中出现的变更、偏差、超标与超常结果严格按对应程序规定进行管控,并在验证报告中予以分析描述。在清洁验证生命周期的3 个阶段中,首先是设计与开发阶段,该阶段涉及的要点最多,需要结合生产线共线生产情况、设备使用情况、原料药清洁级别[5]、共线生产药品性质(包括剂型[6]、活性物质及辅料)开发清洁方法,制定限度标准,开发并验证活性残留物检验方法。通过对设备情况及生产情况分析,确定取样位置及取样方法,进行取样回收率验证。基于对使用后至清洁前的间隔时间、生产最长时间、生产最大批次等内容的评估确定最差条件,根据相似药品及设备的分析明确清洁验证括号设计。其次是验证实施阶段,该阶段在设计与开发阶段工作的基础上,建立对应药品生产设备标准清洁操作规程,制订清洁验证方案,对从事清洁工作的人员进行培训并严格监控。在实施清洁验证过程中注意对设备校准、清洁用具、取样工具的管理,确保验证实施环境与实际生产及存放环境一致,并至少进行3 个批次的验证,最终形成清洁验证报告。最后是持续监测与再验证,在完成清洁验证的实施,确认清洁方法有效后,仍需要进行持续的监测,确保清洁方法的持续有效[7],制定日常清洁监测标准(如目视检查、pH 值、电导率及总有机碳[8]监测等),通过定期确认及周期性的评估确保其有效运行,必要时进行再验证。特殊情况下(如引入新的产品、清洁方法变更等),可能需要重新进行清洁验证。
1.3 药品生产设备清洁验证关键要点分析
在清洁验证要点结构框架中,存在一些较常出现问题及相对较复杂的要点值得重点关注,主要包括:
(1)限度标准:包括目视检查、微生物负荷、活性残留物、清洁剂及消毒剂残留、降解物残留、残留物累积、其他可能的毒性成分残留(如沙坦类药品中的亚硝胺杂质、回收溶剂可能带入的杂质等)的限度标准规定,其中活性残留物的选择是最为核心的内容。活性残留物可以针对每个产品建立,也可从相似品种中选择最差条件的产品作为目标残留物,也可基于溶解性、毒性、活性、清洁难度及可检测情况基于风险选择一种或多种目标残留物,并通过对日常治疗剂量的0.1%、1×10−5 及基于可接受日暴露量(ADE)及允许日暴露量(PDE)等安全性指标计算的限度标准三者中选择最低的值作为活性残留物限度标准[9-10],其中对于中药生产应考虑其特殊性[11]。
(2)取样:包括取样方法,取样位置(基于设备设计与复杂程度)[12]、取样工具、取样人员、取样操作[13]、取样后样品处理、回收率确认、取样设备表面材质等需要关注的内容。取样方法应包括直接取样(棉签擦拭)和淋洗取样,并针对不同的取样位置选择适宜的取样方法,在选择取样位置时需要注意包括最难清洗位置[14],并考虑对连接管道等辅助设备的取样。开展取样方法、取样设备表面材质及取样人员回收率确认(至少大于50%),并注意对取样棉签材质及供应商的管控、取样后样品的处理、现场取样面积模具配备、回收率验证模拟过程与实际情况的一致性等内容。
(3)清洁方法:包括同一产品更换批次的清洁方法(小清)、更换不同产品时的清洁方法(大清)[15]、针对各品种的清洁方法、针对不同设备的清洁方法、清洁程序建立依据、清洁方式(手动清洁、半自动清洁及自动清洁)、具体的清洁程序(如清洁剂、浓度、温度、体积、清洁次数、清洁时间、设备运行参数设置、人员操作、清洁步骤)等。
(4)最差条件:应充分评估生产最长时间、最大批次数、使用后至清洁开始的最长时间间隔、清洁后的存放方式与时间、降解与累积的风险等。
(5)检验方法:活性残留物检测方法必须经过验证,检测限、定量限、线性范围及专属性等应适用于活性残留限度标准,如亚硝胺残留在不同产品中的残留情况检测,不能直接套用美国食品药品监督管理局(FDA)发布的用于检测缬沙坦中是否存在杂质N-亚硝基二甲胺杂质的气相色谱–质谱(GC/MS)顶空法[16]。
2、我国药品检查中药品生产设备清洁验证缺陷的识别和分析
对于存在多产品共线生产的企业,现场检查的重点必然包括清洁验证。通过对国家核查中心2017—2018 年跟踪检查中发现的缺陷进行统计分析,发现涉及清洁验证的缺陷共78 条,主要问题及分布情况见表1。
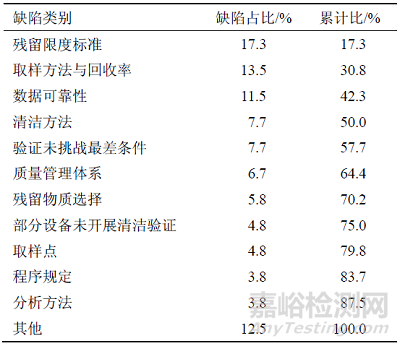
表1 2017—2018 年跟踪检查清洁验证缺陷分布情况
清洁验证的各类缺陷中,居于首位的是残留限度标准的问题,包括残留检验项目不合理(如仅对清洁剂残留进行检测、仅进行目视残留检测、设备碱液浸泡清洗后不测试pH值等),具体限度数值(如紫外吸光度小于特定数值等)制定依据不充分,未按验证方案规定项目进行全项检测等。其次是数据可靠性方面的缺陷,主要包括清洁验证报告及所附清洁记录内容及信息存在缺失或错误(如设备清洁记录未记录清洗方法与过程,遗漏部分设备清洁验证数据、设备型号信息错误等),清洁验证样品检测数据处理不规范(手动积分、禁止积分参数设置等)。其三是部分设备未开展清洁验证,除部分设备没有进行清洁验证外,还存在未对非专用滤芯、链接管道等进行清洁验证的情况。清洁方法的主要问题包括清洁方法未考虑目标物质的溶解性,清洁程序规定(如淋洗用溶媒量)不具体,缺少对手工清洁操作中人员、时间等影响因素的评估。取样方法与回收率方面的问题主要包括取样方法仅使用淋洗法取样(未使用擦拭法),取样规定可操作性差(如对取样面积、取样方式、图示等描述不明确),未对取样人员进行取样回收率验证,取样回收率验证溶液浓度不在可接受范围内等。验证未挑战最差条件的缺陷中,主要是清洁验证中未对更换品种、生产结束至清洁开始的最长时间、最长清洁周期进行验证。取样点的问题集中在最难清洁点选择及已选择取样点的代表性方面。再验证的方面主要是文件中规定的再验证周期缺少合理依据及未按规定进行定期再验证的问题。此外,对于清洁验证中发生的偏差,对清洁验证涉及的共线风险评估报告、分析方法验证及验证批数等方面也发现一些问题的存在。
除在对药品生产企业的GMP 跟踪检查外,在药品注册生产现场核查中,新申报产品引入已有生产线的清洁验证情况也是核查重点,其中包括新产品对原有产品的影响,也包括原有产品对新引入产品的影响。在近几年的注册生产现场核查中,曾出现由于企业共线生产细胞毒性药品的清洁验证不完善,存在污染与交叉污染的风险等问题,导致产品未能获批的情况。
3、境外药品检查中药品生产设备清洁验证缺陷的识别和分析
近年来,基于监管机构间的合作协议,我国药品监督管理部门也会应境外药品检查及监管机构(如世界卫生组织、美国食品药品监督管理局等)的邀请,观察其对我国境内部分药品生产企业的检查。2015—2018 年观察检查中记录的关于清洁验证的缺陷共72 条,见表2。
表2 2015—2018 年境外观察检查中清洁验证缺陷分布情况
与我国药品检查相比,境外药品检查观察在清洁验证方面的缺陷类型基本一致,除各类缺陷的占比存在一些差异外,在缺陷内容方面也存在一些区别,主要包括:
(1)在残留限度标准方面,对微生物检测、1×10−5 标准的合理性、依据ADE 和PDE计算残留限度的要求、TOC 标准与实际残留量的关系、累积与降解情况方面的发现的问题相对较多;
(2)在取样方法与回收率方面,对回收率验证涉及的材质(如密封胶圈、管道、聚丙烯、玻璃等)、擦拭测定中样品干燥条件与实际工艺中干燥条件的一致性、擦拭棉签浸润及浸泡溶剂、取样工具与模具的管理也更为关注;
(3)数据可靠性方面,关注限度标准计算过程的记录及验证方案报告的规范管理;
(4)清洁方法方面,还发现实际清洁操作与验证的清洁方法不一致,存在额外清洁性操作(如清洁后使用有机溶媒进行表面润洗)未纳入验证,不同程序规定的清洁方法不一致,清洁用具管理不规范的问题;
(5)验证未挑战最差条件方面,对最大连续生产批次数进行验证及评估的问题;
(6)质量管理体系方面,主要是清洁验证过程发生的偏差处理、变更控制、超标结果调查中存在不足;
(7)残留物质选择方面,包括活性物质矩阵表缺少对新引入物质的评估、未考虑溶剂残留、选择的活性物质不能代表其他产品等情况;
(8)部分设备未开展清洁验证方面,同时也关注辅助生产设备(如灌装计量枪、滤袋、中转桶)的清洁验证。此外,对于依据欧盟清洁验证规定的要求、残留分析方法验证、清洁验证中的设备分类等方面也发现了一些问题。
通过对2015—2019 年FDA 针对我国境内药品生产企业发出的77 封警告信分析发现,涉及清洁验证缺陷的问题主要包括:伪造清洁验证报告的数据可靠性问题、清洁验证不充分、部分设备未开展清洁验证、未对清洁验证分析检测中出现的异常峰进行调查。
4、结语
科学、有效的药品生产设备清洁验证是避免产生药品污染与交叉污染的重要措施,特别是在多产品共线生产的情况下,直接关系到药品质量与安全,做好药品生产设备的清洁验证对药品生产企业至关重要。在实际工作中,药品生产企业可以参照本文提出的药品生产设备清洁验证要点结构图对清洁验证开展情况进行分析,借鉴国内外药品检查中常见的缺陷进行对照,依据国内外相关法规指南,结合实际情况不断完善、改进清洁验证工作。在药品检查中,药品检查员可以参考清洁验证要点结构图及常见缺陷情况,基于风险开展针对性的检查,进一步提升现场检查质量和效率。
参考文献
[1] 薛 峰. 关于药品GMP 检查中清洁验证常见问题的矫正 [J]. 药学与临床研究, 2020, 28(1): 74-77.
[2] WHO. Good manufacturing practices: guidelines on validation. Appendix 3. Cleaning validation [EB/OL]. [2020-05-03]. https://extranet.who.int/prequal/content/inspections-0.
[3] 翟铁伟. 药品生产中清洁验证的生命周期探讨 [J]. 中国医药工业杂志, 2019, 50(11): 1341-1347.
[4] 姜 彬. 质量风险管理在非无菌原料药清洁验证评估中的应用 [J]. 中国药师, 2017, 20(12): 2281-2285.
[5] APIC. Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants [EB/OL].(2016-09-12) [2020-05-03].
[6] 郝莹华, 梁 毅. 原料药生产设备清洁验证的范围和程度评估 [J]. 中国医药工业杂志, 2018, 49(4): 522-527.
[7] 熊 浪, 梁 毅. 基于生命周期理论的共线生产清洁验证关键点研究 [J]. 制药装备, 2016, 12(12): 8-12.
[8] 庄目德. 清洁验证TOC(总有机碳)取样回收率研究[J]. 海峡药学, 2017, 29(7): 73-75.
[9] EMA. Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities [EB/OL].(2014-12-20) [2020-05-05].
[10] EMA. Questions and answers on implementation of riskbased prevention of cross-contamination in production and guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities [EB/OL]. (2018-04-19) [2020-05-05].
[11] 王守斌, 肖传学, 黄雪红, 等. 多品种共线的中药制剂清洁验证 [J]. 中草药, 2016, 47(10): 1815-1819.
[12] 柴振平, 高 鹏, 白亚灵, 等. 化学药片剂生产设备清洁方法的验证 [J]. 中国药房, 2015, 26(34): 4755-4758.
[13] 马肖梦, 黄丽敏, 许汉林. 关于设备清洁残留物限度验证的探讨 [J]. 湖北中医杂志, 2016, 38(8): 69-72.
[14] 高 歌, 张会云, 尤春玲, 等. 制药企业共线生产产品清洁验证 [J]. 质量探索, 2016, 138(4): 46-47.
[15] 王守斌, 聂 杰, 陈如柳, 等. 浅析药品生产设备的清洁验证 [J]. 天津药学, 2014, 26(5): 72-76.
[16] FDA. FDA-published testing method to provide an option for regulators and industry to detect NDMA impurities[EB/OL]. (2019-10-17) [2020-05-04].