您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-06-02 13:51
摘要:通过梳理美国FDA发布的34个吸入制剂仿制药开发特定药品指导原则,总结了吸入粉雾剂、吸入气雾剂、吸入喷雾剂、吸入混悬液和吸入溶液仿制药开发所用的体外生物等效性研究、药动学研究、药效学研究、临床终点研究和装置比较方法,比较了各品种开发方法、试验设计、主要研究终点和等效性标准方面的差异,为吸入制剂仿制药开发提供更有针对性的参考。
2020年美国FDA发布的仿制药开发特定药品指导原则(Product-specific Guidances for Generic Drug Development,PSG) 为仿制药产品提供了最合适的开发方法和获批所需支持性证据的要求,截至目前已发布1865个指导原则,大大促进了仿制药开发、申请和批准,增加了民众获得安全、廉价仿制药的可能性 [1]。近年来,我国吸入制剂仿制药开发和申报数量呈快速增长趋势,如何确定吸入制剂仿制药的开发方法和生物等效的标准是行业关注的焦点。2020年12月15日,国家药品监督管理局药品审评中心 (CDE) 发布了《经口吸入制剂仿制药生物等效性研究指导原则》,明确了经口吸入制剂仿制药生物等效性 (bioequivalence,BE) 研究的一般性原则 [2],为业界提供参考和依据。由于吸入制剂仿制药BE研究的复杂性,不同剂型和品种可能需要不同的方法和策略,因此参考具体品种的指导原则可能更有针对性。
FDA发布的仿制药开发特定药品指导原则中有34个涉及吸入制剂,这些指导原则推荐的仿制药开发方法通常包含体外BE研究、药动学研究、药效学研究、临床终点研究和装置比较方法,涉及的剂型有吸入粉雾剂、吸入气雾剂、吸入喷雾剂、吸入混悬液和吸入溶液。本文将介绍此34个吸入制剂的特定药品指导原则并进行梳理,为吸入制剂仿制药开发提供有针对性的参考。
1品种信息介绍
34个经吸入途径给药的特定药品指导原则涉及的适应证,主要包括慢性阻塞性肺疾病 ( chronic obstructive pulmonary disease,COPD)或(和)哮喘、可逆性阻塞性气道疾病和预防恶性胸腔积液复发,活性成分为支气管扩张药、激素类药物和矿物质药物,剂型有吸入粉雾剂 (18个)、吸入气雾剂(含胸膜内吸入气雾剂,下同,13个)、吸入喷雾剂 (1个)、吸入混悬液 (1个) 和吸入溶液 (1个)。其中吸入粉雾剂和吸入气雾剂数量最多,PSG推荐体外BE研究、药动学研究、药效学研究或临床终点研究和装置比较方法;吸入喷雾剂和吸入混悬液,PSG仅推荐体外BE研究和药动学研究方法 ;吸入溶液,PSG推荐豁免体内研究方法。具体品种信息见表 1。
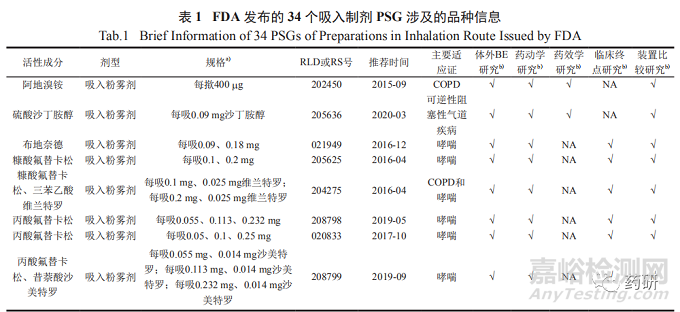
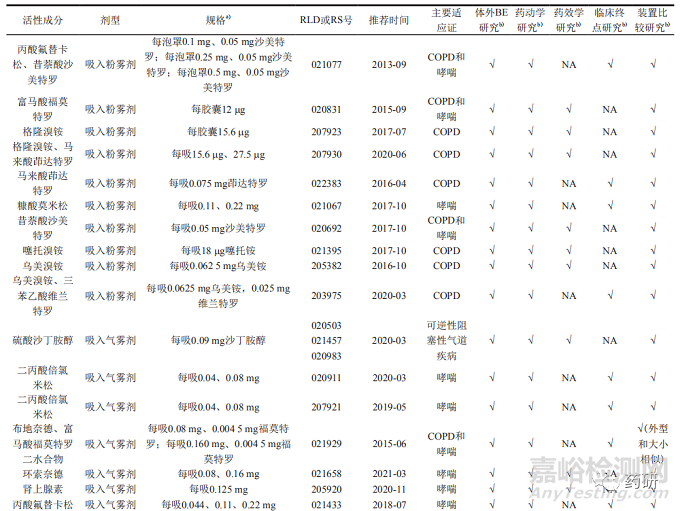
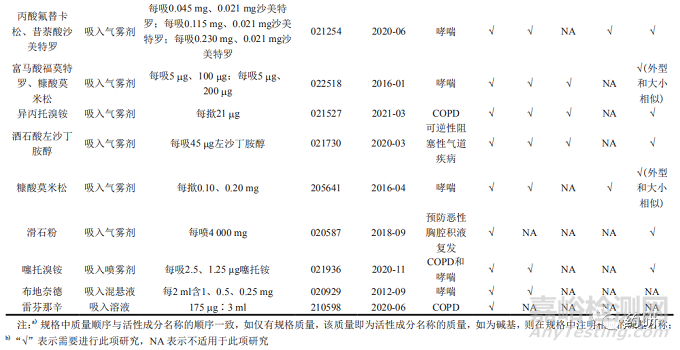
2体外BE研究
FDA建议申请人对受试制剂和参比制剂的所有规格进行以下体外BE研究。每个规格至少使用3批受试制剂和参比制剂,每批不少于10个样品。FDA建议3个主要稳定性批次也可用来证明体外BE。3批受试制剂应至少由3批不同的原料药、辅料和装置组件或包装系统制备。受试制剂应由拟上市的最终装置组件和最终处方组成。通常建议受试制剂与参比制剂的非活性成分类型 (Q1) 和用量(Q2) 相同,如果受试制剂与参比制剂Q2不同,应说明原因,并且应提供药物开发数据,如活性成分与赋形剂多个组合配比的体外试验 ( 包括低于或高于参比制剂所用活性成分与赋形剂比率的组合配比) [3]。PSG对吸入粉雾剂、吸入气雾剂、吸入喷雾剂、吸入混悬液和吸入溶液仿制药的体外 BE 研究要求不同,具体如下。
2.1 吸入粉雾剂
2.1.1单揿含量 (single actuation content,SAC)
SAC试验应在产品生命周期的初始、中间和最后阶段测定,流速为30、60和90L/min。采用美国药典 (USP)<601> 仪器B或其他经验证的适当方法测定。每次测定使用的胶囊数量应为1个,每个胶囊测定所用的揿次应为1次。递送系统抽取的空气量应为2L。
等效性标准 :SAC的群体生物等效性(population bioequivalence,PBE) 分析。关于PBE分析步骤的信息可参考布地奈德吸入混悬液的PSG[3]。
2.1.2空气动力学粒径分布(aerodynamic particle size distribution,APSD)
APSD 试验应在产品的初始和最后阶段测定,流速为28.3或30、60和90L/min。可使用USP<601> 仪器3、仪器5或其他经验证的适当方法测定。每个剂量的APSD测定应使用最小的胶囊数量,并证明已验证方法的灵敏度。递送系统抽取的空气量应为4L。
附加说明 :需计算各层级的沉积量 ( 包括吸嘴适配器、导入口、预分离器以及级联撞击器和过滤器 )。应根据所有层级沉积量的总和报告质量平衡组成。
等效性标准 :撞击器内质量 ( impactor-sized mass,ISM) 的PBE分析。应提交级联撞击器各层级沉积量的分布曲线,质量中位数空气动力学直径(mass median aerodynamic diameter,MMAD)、几何标准差 (geometric standard deviation,GSD) 和微细粒子剂量(fine particle mass,FPM),作为APSD等效的支持性证据 [3]。
2.2 吸入气雾剂
吸入气雾剂除了进行SAC和APSD测定外,还需进行喷雾模式、羽流几何形状、填充和再填充测定。
2.2.1喷雾模式
喷雾模式应在产品的初始阶段和距驱动器孔的2个不同距离处进行测定。选定的距离应至少相距3cm,并距参比制剂驱动器吸嘴部位3 ~ 7cm。可用碰撞 (薄层色谱板嵌塞 )、非碰撞 (激光光片技术 )或其他合适的方法测定。
附加说明 :应根据自动分析的椭圆度比 (定义为Dmax 与Dmin 的比值,其中Dmax 和Dmin 分别是通过质量中心或重心的最长和最短直径 ) 和真实形状周长内面积 ( 包括高比例,例如总喷雾模式的95% ) 或手动分析的椭圆度比和Dmax 定量测定。每个喷雾模式的喷雾次数最好是1次。
等效性标准 :在2个选定的距离对以下2方面进行分析。①喷雾形状的定性比较 ;②椭圆度比和真实形状周长内面积或椭圆度比和Dmax的PBE分析 [4]。
2.2.2羽流几何形状
羽流几何形状应在产品生命周期的初始阶段测定。定时序列声触闪光照相法、激光光片技术或其他合适的方法均可用于测定揿后延迟适当时间的羽流几何形状。
附加说明 :当全部喷出的羽流几何形状仍与驱动器吸嘴接触时,报告在单个延迟时间测得的羽流几何形状。羽流几何形状应根据羽流角度和宽度进行定量测量。羽流角度是在驱动器吸嘴部位或附近的角,并延伸出羽状的锥形区域。测定羽流宽度的距离等于所选喷雾模式的较大距离。
等效性标准 :3批受试制剂和参比制剂羽流角度和宽度的几何平均值的比值 ( 经对数转换数据 )应在90%~ 111% [4]。
2.2.3填充和再填充
填充和再填充试验应考虑在参比制剂标签中规定的填充和再填充次数之后立即进行单揿递送剂量测试 ( 在驱动器以外 )。如果参比制剂标签提供了再填充的信息,则再填充试验应在产品初次使用后贮存指定的非使用时间和 ( 或 ) 在其他条件下测定。
附加说明 :BE评估建议气雾剂以阀门直立位置贮存进行填充和再填充试验,但参比制剂标签建议气雾剂以阀门倒立位置贮存的情形除外。填充数据可基于初始阶段的SAC数据。
等效性标准 :在参比制剂标签中规定的填充或再填充揿数之后,立即对单揿的递送剂量进行PBE分析。
无菌滑石粉吸入气雾剂的体外研究项目包括递送剂量、递送速率、喷雾模式和羽状几何形状 [5]。
2.3 吸入混悬液
FDA发布的吸入混悬液PSG仅有布地奈德吸入混悬液,该指导原则的体外研究分为高规格和低规格,具体如下。
2.3.1 1mg∶2ml 规格的体外BE研究要求
布地奈德吸入混悬液仿制药中非活性成分的Q1和Q2必须与参比制剂相同。建议使用Pari LC Plus 雾化器 /Pari 主压缩机系统进行雾化。测定项目包括以下7项。
①药物晶型相同 (X 射线衍射法 )。
② 药物的晶体外形 ( 晶癖 ) 相同。
③ 比较安瓿中药物的单位剂量含量 (unit dose content,UDC)。
④ 比较平均雾化时间 (mean nebulization time,MNT)和平均递送剂量(mean delivered dose,MDD):吸嘴处应以5.5L/min 的标示流速进行试验,直至雾气不再从吸嘴处流出。
⑤ 比较混悬液 ( 安瓿 ) 中的药物颗粒和附聚物的粒度分布 (particle size distribution,PSD) :PSD的测定应采用经验证的方法 ;验证应证明方法对混悬液中预期粒径范围内药物粒径的灵敏度。
⑥ 比较雾化气溶胶中药物颗粒和聚集物的PSD :推荐采用USP<601> 装置5,在流速为15L/min 条件下雾化气溶胶,进行APSD的测定。建议照 USP<1601> 采用Pari LC Plus 雾化器/Pari主压缩机系统进行研究。应提交导入口、级联撞击器的7个层级、备用过滤器和微孔收集器 (microorifice collector,MOC) 中药物沉积量的总和。
⑦ 比较激光衍射法测定的雾化气溶胶的液滴粒度分布 [6]。
2.3.2 0.5或0.25mg∶2ml 规格的体外BE 研究要求
如果高、低规格制剂使用的微粉化布地奈德 (原料药 ) 相同,即相同的粒径、PSD、多晶型和晶癖,并且与参比制剂低规格的Q1和Q2相同,建议对低规格制剂进行体外BE研究。
如果可以测定高、低规格受试制剂和参比制剂的雾化气溶胶中药物颗粒和聚集物的PSD,则以下体外试验应该能够证明低规格制剂的等效性。
① 根据上述可接受的体外比较数据,高规格制剂生物等效性的文件。
② 比较低规格受试制剂和参比制剂混悬液 ( 安瓿 ) 中的药物颗粒和聚集物的PSD。
③ 比较低规格受试制剂和参比制剂的雾化气溶胶中药物颗粒和聚集物的PSD。
④ 比较低规格受试制剂和参比制剂药物的UDC。
⑤ 比较MNT和MDD。
⑥ 高、低规格受试制剂的MDD 比值应与参比制剂相似 [6]。
2.3.3留样数量
① 如果在一个场地进行体内外BE研究,受试制剂和参比制剂 (包括安慰剂) 每批至少保留50个样品,多剂量制剂每罐或每瓶有30 揿以上。
② 如果在多个场地进行体内外BE 研究,受试制剂和参比制剂 (包括安慰剂 ) 每批至少保留50个样品,每个场地不少于10个样品 [6]。
2.4 吸入喷雾剂
FDA发布的吸入喷雾剂PSG仅有噻托溴铵吸入喷雾剂,体外BE研究需按照气雾剂体外BE研究测定SAC、APSD、喷雾模式、羽流几何形状、填充和再填充,另外还需要研究喷雾持续时间和速度。
2.4.1喷雾持续时间
喷雾持续时间应在产品生命周期的初始和最后阶段测定,可使用高速摄像机录像、激光衍射、粒子图像测速或其他合适的方法测定。
等效性标准 :对从喷雾开始喷出,到在喷嘴形成喷雾最后时刻的时间间隔进行PBE或其他适当的统计分析。如果使用其他统计分析,则应充分考虑研究目的并具有科学合理性[7]。
2.4.2喷雾速度
喷雾速度应在产品生命周期的初始和最后阶段测定,可用高速成像、粒子图像测速、相位多普勒或其他合适的方法测定。
等效性标准 :在距喷嘴8 ~12cm处对羽流前缘速度进行 PBE 分析或其他适当的统计分析。如果使用其他统计方法,则应充分考虑研究目的并具有科学合理性,应提交完整的羽流前缘速度与距离数据作为喷雾速度等效的支持性证据 [7]。
2.5 吸入溶液
FDA发布的吸入溶液PSG仅有雷芬那辛吸入溶液,指导原则建议当受试制剂和参比制剂有相同剂型和浓度,且不含有显著影响系统或局部生物利用度的非活性成分时,可以豁免体内研究。如果受试制剂和参比制剂Q1和Q2不同,应对至少3批受试制剂和参比制剂的相关质量和性能属性进行表征,包括外观、pH 值、渗透压和其他任何潜在相关的物理和化学性质 [8]。
3 药动学研究
吸入粉雾剂、吸入气雾剂和吸入喷雾剂PSG对于药动学BE研究的要求基本相同,建议所有受试制剂和参比制剂的规格均应进行药动学研究,一般采用空腹、单剂量、双交叉的试验设计,受试者选取健康男性和未妊娠女性的一般人群,分析物为原型药,剂量采用在灵敏分析方法下足以表征药动学曲线的最小吸入次数 [3]。
附加说明 :①参与体内研究的受试者,应在每次治疗前接受标准方式的吸入制剂使用培训,以确保相对一致的吸气流速和吸气持续时间。②如果剂量超过最大标示单次剂量,则应在药动学研究之前进行研究性新药申报[3]。③可采用参比制剂标度的平均BE。应提供BE参数、AUC和(或)cmax高变异的证据 (即受试者个体内变异系数≥30%)[9]。
等效性标准 :通常采用AUC和cmax,受试制剂和参比制剂的AUC和cmax的几何均值比的90%置信区间应在80%~125%。值得注意的是硫酸沙丁胺醇气雾剂BE研究有AUC0 → t、AUC0 → ∞ 和cmax3 个指标[4]。硫酸沙丁胺醇吸入粉雾剂和吸入气雾剂、酒石酸左沙丁胺醇吸入气雾剂的给药剂量分别为 0.18mg(2 吸)、0.18mg(2 吸)和 0.09mg(2 吸)。无菌滑石粉吸入气雾剂未推荐药动学研究方法 [5]。布地奈德吸入混悬液仅1mg ∶2ml 规格推荐了药动学研究方法,但未明确药动学研究的具体内容 [6]。
4药效学研究
药效学研究一般采用随机、单剂量、安慰剂对照、交叉或平行研究,2周导入期。受试者为非生育或节育的成年男性和女性哮喘或COPD患者 ;如参比制剂适应证包含哮喘和COPD,则选用哮喘患者。给药剂量一般为单剂量,每次1吸。主要终点包括 :治疗后0 ~t h,1秒内用力呼气量 (FEV1)-时间曲线下面积 (AUC0 → t),根据药效的作用时长,t 可为4、6、12或24 h ;4或8周治疗期最后一天给药前早晨的FEV1 ;对于部分品种的每个治疗组,每个评估期内所有测定时间点的支气管扩张药反应峰值时间 ( tmax) 和FEV1值应包括在最终研究报告中。研究终点应进行基线调整。为保证研究具有充分的敏感性,受试制剂与参比制剂的主要研究终点应在统计学上优于安慰剂 (P<0.05)。等效性标准通常为 :受试制剂与参比制剂主要终点比值的90%置信区间应在80%~125%。其中富马酸福莫特罗和糠酸莫米松吸入气雾剂给药剂量为多剂量,每日 2次,每次2吸 [10] ;噻托溴铵吸入粉雾剂的给药剂量为单剂量,每次2吸 [11]。
硫酸沙丁胺醇吸入粉雾剂和吸入气雾剂,以及酒石酸左沙丁胺醇吸入气雾剂 PSG推荐使用支气管激发研究方法。试验设计为单剂量、双盲、双模拟、随机、交叉研究。4组给药剂量分别为:① 零剂量:2个不同的参比制剂安慰剂,各1次1吸 ;2个不同的受试制剂安慰剂,各1次1吸。② 0.09mg 参比制剂:参比制剂和参比制剂安慰剂,各1次1吸;2个不同的受试制剂安慰剂,各1次1吸。③ 0.18mg参比制剂 :2个不同的参比制剂,各1次1吸 ;2个不同的受试制剂安慰剂,各1次1吸。④ 0.09mg受试制剂 :受试制剂和受试制剂安慰剂,各1次1吸 ;2个不同的参比制剂安慰剂,各1次1吸。每组治疗之间的清洗期不少于 24h。受试者选择非生育或节育的成年男性和女性稳定轻度哮喘患者。药效学终点为给药后FEV1降低20%所需激发剂的浓度 (PC20) 或剂量 (PD20),即吸入不同剂量的沙丁胺醇 ( 或安慰剂 ) 后,FEV1降低20%所需的乙酰甲胆碱激发剂的激发浓度或剂量。FEV1降低20%是相对于服用安慰剂或沙丁胺醇前用生理盐水测得的FEV1值。
等效性标准 :药效学数据的剂量标度分析。剂量标度分析的详细信息请参考奥利司他口服胶囊的PSG。相对生物利用度 (F) 的90%置信区间应在67%~ 150% ( 由于药效学测量指标的变异性通常大于药动学测量指标的变异性,因此等效性评价的置信区间放宽到67%~ 150% )。需要注意的是乙酰甲胆碱对气道反应性的结果应说明基线PC20 ≤8 mg/ml( 服用沙丁安醇前 )。基线 FEV1 不应低于预测正常值的70%,并应在确认当天FEV1 的88%~112%,如 FEV1 不在上述范围,应重新研究。基线FEV1相比生理盐水对照引起的 FEV1 下降应不超过10%,否则应推迟研究。在进行药效学研究前,需要进行研究性新药申报,因为氯化乙酰甲胆碱溶液的浓度可能超过标示的25.0 mg/ml,特别是在较高剂量的沙丁胺醇 ( 例如0.18mg) 条件下,25.0mg/ml的氯化乙酰甲胆碱可能不会导致FEV1降低20%。FDA鼓励企业考虑开展预试验,以完善研究设计( 如入排标准 ),并根据受试者个体内和个体间的变异性和最大效应剂量 - 反应曲线的斜率估计研究效力。
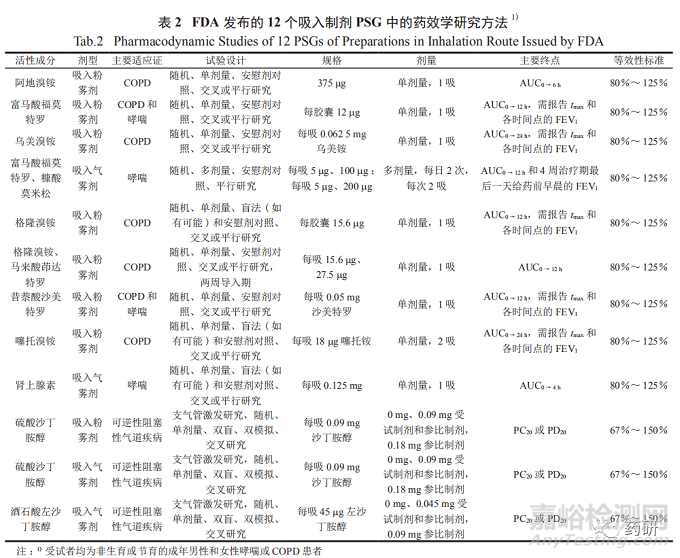
FDA 发布的吸入制剂PSG中有12个含药效学研究方法,简要信息汇总见表 2。
5临床终点研究
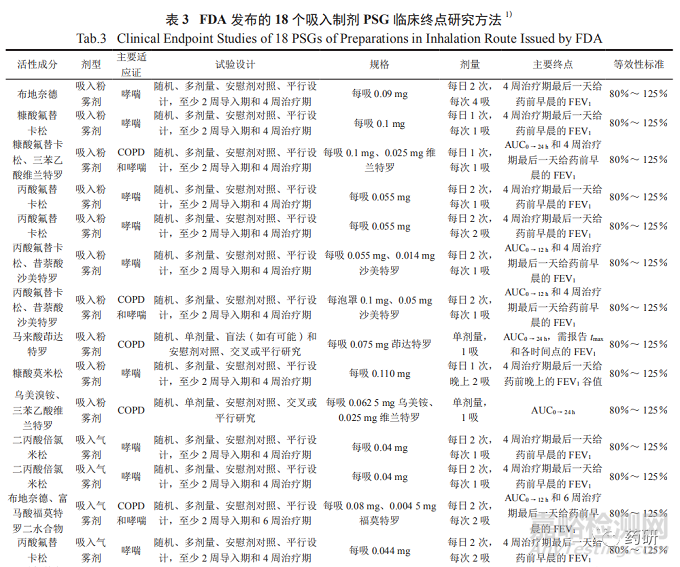
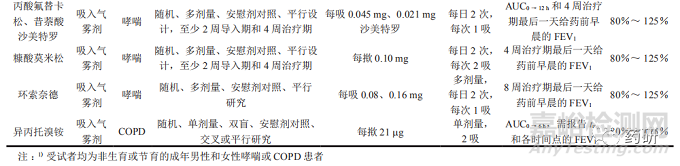
对于多规格的制剂,FDA发布的PSG显示采用临床终点研究方法的品种均推荐采用最低规格进行研究,通常选择最低规格能够更灵敏地反映临床终点指标。一般采用随机、多剂量、安慰剂对照、平行设计,至少2周导入期和4周治疗期的试验设计。受试者选择非生育或节育的成年男性和女性哮喘或COPD患者,诊断为哮喘至少12个月。给药剂量一般为每日2次,每次1吸或2吸。主要终点为 4周治疗期最后一天给药前早晨的FEV1和(或)AUC0 → t。主要终点应进行基线调整 ( 相对于基线的变化 )。FEV1基线是指在4周治疗期的第一天早晨,测量给药前至少2个时间点的FEV1的平均值。建议采样时间与4周治疗期最后一天的时间相同。
等效性标准 :受试制剂与参比制剂主要终点比值的90%置信区间应在80%~125%。其中布地奈德和富马酸福莫特罗二水合物吸入气雾剂PSG推荐6周治疗期的试验设计,主要终点为AUC0 →12 h和6周治疗期最后一天给药前早晨的 FEV1[12]。乌美溴铵和三苯乙酸维兰特罗吸入粉雾剂,PSG推荐采用随机、单剂量、安慰剂对照、交叉或平行研究的试验设计,受试者选择非生育或节育的成年男性和女性COPD患者,给药剂量为1吸,主要终点为AUC0 →24h [13]。
二丙酸倍氯米松、环索奈德和异丙托溴铵吸入气雾剂的PSG中提出了临床终点BE替代方法的建议,简要介绍了附加支持性体外研究的方法,指出受试制剂不是水相的处方,而是液化抛射剂的处方,揿后会快速挥发。因此,到达肺部局部作用部位的药物形式不是含有药物溶液的液滴,而是非挥发性的残留药物颗粒,由于呼吸道中相对湿度较高,残留药物颗粒具有复杂的形态。在这种情况下,考虑到PSG已推荐的现有体外和体内药动学BE研究,受试制剂和参比制剂的比较临床终点BE研究是目前能提供肺部局部作用部位临床效果等效性信息的唯一工具。为支持开发新的BE方法和简化的新药申请 (abbreviated new drug application,ANDA),FDA仿制药办公室 (Office of Generic Drugs,OGD)建议应用一个全面的、重要的数据体系进行科学论证,并尽可能进行有统计意义的评估,这可能包括体外、体内和 ( 或 ) 计算机研究方法。对于溶液处方的特定药品,如果受试制剂与参比制剂有相同的Q1和Q2,并且受试制剂装置与参比制剂装置的关键设计属性和用户界面足够相似,则附加的支持性数据可为确保等效性提供依据。因此,在证据权重法的条件下,受试制剂与参比制剂在肺部局部作用部位的检测可被视为当前推荐的比较临床终点BE研究方法的潜在替代方法 [14]。
附加支持性体外研究方法包括但不限于 :① 使用具有代表性的口-喉模型和呼吸曲线进行更具预测性的APSD试验 ;② 根据速度曲线和蒸发率对递送的气溶胶飞沫进行表征 ;③ 溶出度试验 ;④ 形态成像比较,包括表征全部粒径范围的残留药物。潜在申请人还可以考虑使用定量方法、建模 ( 例如基于生理学的药动学和计算机流体动力学研究 ) 和替代体内药动学BE研究的方法。其中异丙托溴铵吸入气雾剂的PSG中指出,还可以考虑评估非挥发性辅料对挥发性辅料的蒸发速率和程度及其沉积在肺部的最终状态 ( 例如干颗粒、半干颗粒、液滴 )的影响。为了在产品开发的早期说明FDA对潜在申请人的期望,并帮助申请人提交尽可能完整的ANDA,FDA极其鼓励申请人通过预先ANDA会议途径,讨论获得替代方法的开发计划[14—16]。
FDA 发布的吸入制剂PSG中有18个含临床终点研究方法,简要信息汇总见表 3。
6装置对比
PSG中鼓励申请人在提交ANDA之前向OGD提交装置的工作模型和工程图纸。潜在申请人可参考FDA指南《药品器械组合产品的比较分析和人为因素研究相关的使用比较》( 2017年1月 ),该指南体现了FDA当前对识别和评估拟定仿制器械组合产品的用户界面,以及与参比制剂设计差异的考量 [3]。
6.1吸入粉雾
剂通过汇总FDA发布的吸入粉雾剂PSG中关于装置相似性的要求,总体上考虑以下几方面特征 :被动 ( 呼吸驱动 ) 装置 ;多剂量定量装置模式或预定量单剂量胶囊模式 ;与参比制剂剂量相同 ;与参比制剂的外部操作步骤和关键设计属性相似 ;与参比制剂装置尺寸和外型相似 ;与参比制剂装置的阻力可比 ;剂量指示器或计数器 ;与参比制剂的患者反馈机制相似。此外,还应进行体外和使用研究支持受试制剂的功能性、准确性和耐用性。
6.2吸入气雾剂
通过汇总FDA发布的吸入气雾剂PSG中关于装置相似性的要求,总体上考虑以下几方面特征 :参比制剂装置尺寸和外型 ;参比制剂的剂量数 ;参比制剂的外部操作步骤和关键设计属性 ;剂量指示器或计数器。此外,还应进行体外和使用研究支持受试制剂的功能性、准确性和耐用性。二丙酸倍氯米松吸入气雾剂,PSG中建议申请人提供数据,证明受试制剂符合标签要求 :①使用前不需要填充和再填充 ;②20L/min 的吸气流速能够触发1揿 [14]。
6.3吸入喷雾剂
考虑以下几方面特征 :主动吸入、定量、多剂量装置 ;参比制剂装置尺寸和外型 ;参比制剂的剂量 ;与参比制剂的外部操作步骤和关键设计属性相似 ;剂量指示器或计数器 [7]。
布地奈德吸入混悬液和雷芬那辛吸入溶液未对装置有明确要求 [6,8]。
7总结
FDA发布的吸入制剂PSG明确了仿制药开发的方法和支持批准所需的证据,总体上吸入粉雾剂和气雾剂均需要体外、体内研究以及装置对比来证明受试制剂与参比制剂的等效性,多数支气管扩张药物的体内研究需要进行药动学和药效学研究,激素类药物的体内研究则需要进行药动学和临床终点研究。对于布地奈德吸入混悬液,FDA认为布地奈德不溶解,辅料溶解在混悬液中,如果仿制药中的布地奈德和辅料与原研药相同,其微粒特性是仅有的潜在差异,如果布地奈德的微粒大小等同,仿制药与原研药在作用部位应有相同的吸收程度和速度,布地奈德经肺吸收进入体循环的量应是等效的,且体外研究方法比临床终点方法更敏感,因此未推荐布地奈德吸入混悬液的体内研究方法。由于雷芬那辛吸入溶液和噻托溴铵吸入喷雾剂均为溶液剂,在辅料不影响吸收的条件下,药物本身的吸收是没有差异的,因此均推荐豁免体内研究。无菌滑石粉吸入气雾剂通过注入胸腔,堵塞胸膜间隙,防止胸膜液再积聚,用于患者恶性胸腔积液的复发,从作用机制看滑石粉并不通过吸收至体内发挥作用,因此PSG仅推荐了体外研究策略。
采用药效学研究方法进行等效性评价的药物主要是支气管扩张药,主要终点指标是AUC0 →t,也有激素类药物采用药效学研究方法,如环索奈德吸入气雾剂、富马酸福莫特罗和糠酸莫米松吸入气雾剂,单方的主要终点指标为肺功能指标 FEV1,复方的主要终点指标为FEV1和AUC0 →t。硫酸沙丁胺醇类吸入气雾剂采用支气管激发研究方法进行等效性评价,由于方法本身有较大的变异性,因此等效性评价的置信区间也相应放宽。
采用临床终点研究方法进行等效性评价的药物主要是激素类药物,主要终点指标是肺功能指标FEV1,也有支气管扩张药采用临床终点研究方法,如马来酸茚达特罗吸入粉雾剂、乌美溴铵和三苯乙酸维兰特罗吸入粉雾剂,主要终点指标均为AUC0 →t。值得注意的是糠酸莫米松吸入粉雾剂等效性评价的主要终点为4周治疗期最后一天给药前晚上的FEV1谷值,这与糠酸莫米松吸入粉雾剂原研品种的服药方式有关,该品种原研药的说明书要求当每天1次服药时,只能在晚上服药 [17]。
二丙酸倍氯米松、环索奈德和异丙托溴铵吸入气雾剂的PSG中均提出临床终点BE替代方法的建议,并简要介绍了附加支持性体外研究方法,这些方法不仅可用于上述3个品种的体外研究,也可根据品种的特点用于评价其他吸入气雾剂品种,为吸入气雾剂仿制药的开发提供了新的思路。
2020年底,CDE发布了《经口吸入制剂仿制药生物等效性研究指导原则》,明确了经口吸入制剂仿制药BE的一般性原则,与FDA发布的吸入制剂PSG有较多的相似之处,仿制药企业可根据国内指导原则的要求进行开发,具体的品种也可参考FDA发布的PSG更有针对性地进行开发。由于吸入制剂仿制药开发和BE 评价的复杂性,建议对于开发中关键的技术问题,企业应积极与审评机构沟通,以节省开发时间和成本。

来源:Internet


