您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-04-05 20:53
ICH Q3A最新版本距今快15年了,具体内容和要求,已经很清晰明朗了。
作为一名原料药工艺开发人员,小编再聊一下这个指南。
小编觉得ICH Q3A的精髓是杂质阈值的规定,和实际工作息息相关。
附件1:精髓部分
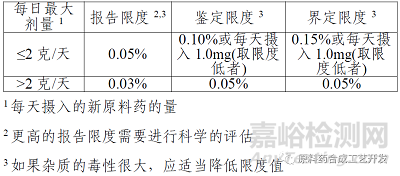
如果原料药最大日剂量不超过2g,报告限度0.05%;如果最大日剂量是0.9g,某杂质A含量0.10%,0.10%*0.9g=0.9mg低于1.0mg,则鉴定限以0.10%为界;如果杂质A含量0.15%,0.15%*0.9=1.35mg高于1.0mg,则定量限不能以0.15%为界,应该以不得过1.0mg为界,含量0.15%的杂质A应该经过纯化等手段降低到不超过1.0mg/0.9g=0.11%。
如果原料药最大日剂量超过2g,直接执行表格要求就行。
题目:“新”“原料药”中的杂质
指导原则是针对原料药的,不是起始原料和中间体,所以起始原料和中间体中的杂质不一定要完全按照本指导原则中的杂质限度进行控制。只要杂质来源,去向,对中间体或者原料药的质量影响有了充分研究,杂质可以是已知的,可以是未知的,可以0.10%,可以0.15%,还可以0.5%或者更高。
指导原则是针对新原料药的,何为新?未上市的为新。
仿制药中杂质谱,根据工艺具体信息制定,不同路线的杂质谱不一样。仿制药中杂质限度,要根据各国药典,进口注册标准,ICH Q3A,参比制剂等信息综合制定。不一定要按照ICH Q3A制定,有的已知杂质可以超过0.15%的定量限。
内容:杂质分类
ICH Q3A文件对杂质的大类和小类均进行了概括,这里不具体聊。
实际工作中,ICH Q3A指南更倾向于是有关物质的指导文件,因为残留溶剂有ICH Q3C,金属等残留有ICH Q3D,致突变杂质有ICH M7,无机杂质有药典或者进口注册标准等。
需要注意的是,ICH Q3A关于有机杂质(小编理解为有关物质)分类,包括已知杂质和未知杂质,未知杂质又包括特定的和非特定的。
还需注意,ICH Q3A文件的杂质分类不包括对映异构体杂质,平时工作中,对映异构体一般采用正相方法独立控制。
内容:杂质报告和限度
工艺过程,所有实际存在的和最可能产生的潜在杂质(包括降解杂质)需要进行综述。这里的“最可能”用的很好,充分思考空间…
不可能把反应液中所有杂质弄清楚,甚至分离的中间体中都不可能把所有杂质研究清楚,所以实际存在的和最可能产生的潜在杂质是个综合评估和讨论的工作,哪些杂质在起始原料中研究,哪些在中间体研究,哪些在原料药中研究。
申报资料中,要对含量大于鉴定限度的杂质进行结构确认,没有特殊数据支持,单个杂质不能过定量限。当某个杂质结构无法确认时,申报资料中也应该提供对该杂质所进行的不成功的研究工作的总结,该杂质限度不得过鉴定限度。通常没有必要对表观含量在鉴定限度以下的杂质进行鉴定,但是必须评估这些杂质不是特殊杂质,例如致突变杂质。
允许有非特定未知杂质,允许有特定未知杂质,如果不能鉴定结构,提供合理的研究数据,也是申报加分工作,但不管如何不能超过鉴定限。
内容:杂质报告内容
批次产品分析结果要数字化,不应该用符合规定,符合限度的一般性术语。工作中我们是怎么做的呢?是不是也写成了符合规定?
批次产品,应报告检测到的大于报告限度的任何杂质和总杂质的含量。
所有大于报告限度的杂质应该进行累加,并作为“总杂质”予以报告。不同企业中,“总杂”做法很多,有的是“其他总杂”,有的100%-主峰%的总杂。
在新原料药质量标准中收载的杂质,应该根据在采用拟上市工艺生产的批次中所发现的杂质来加以取舍。质量标准收紧容易放宽难,研发期间,分析方法验证的质量标准应该充分,多充分,充分思考空间...
未鉴定的杂质以相对保留时间标识。相对保留时间是一个值,不是范围;为避免分析方法转移中峰漂移导致相对保留时间对不上,请提前思考解决方法。

来源:原料药合成工艺开发


