您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-04-30 09:17
复杂注射剂具有技术壁垒高、临床优势明显、市场表现良好、可以延长产品生命周期等特点,一直备受关注。本文就复杂注射剂的特点、在美欧日的注册分类、美欧日对复杂注射剂非临床和临床研究的要求及批准案例进行了介绍。
我国现有法规和指南并无特殊注射剂或复杂注射剂的统一定义,2018年1月发布的《化学药品注射剂基本技术要求(试行)》中“特殊注射剂”是指制剂因素可能影响药物体内药代动力学行为的制剂,例如脂质体、微球、微乳等[1]。美国食品药品监督管理局(FDA)“复杂产品”中涉及到的注射剂有脂质体、注射用混悬剂、长循环注射剂、长循环植入剂、自注射复杂药械组合产品等[2]。本文所述的复杂注射剂是一类复杂的载药系统,包括微球、脂质体、微乳、纳米粒(纳米晶和载药纳米粒)、植入体等。
根据是否有同品种上市,复杂注射剂可以分为新药和仿制药2种情况。对于新药而言,又可根据其活性成分是否已在其他已上市制剂中使用分为3种情况:一是该化合物作为新分子实体上市,如月桂酰阿立哌唑长效注射剂;二是该化合物首次上市时的剂型是普通剂型,后来又有复杂注射剂上市,例如利培酮,首次上市的剂型为利培酮普通片剂和口服液,此后上市的是2周给药1次的注射用利培酮微球;三是已有复杂注射剂上市,后续有更长周期的复杂注射剂上市,比如棕榈酸帕利哌酮长效注射剂,首先上市的为1个月周期的制剂,此后上市的为3个月周期的制剂。后2种情况按照我国的新的化学药品注册分类,可以划入到改良型新药的范畴。新分子实体的复杂注射剂开发难度太大,对于我国企业来说,主要以改良型新药和仿制药的开发为主。本文综述了美国FDA、欧洲药品管理局(European Medicines Agency,EMA) 和日本独立行政法人医药品医疗器械综合机构(Pharmaceuticaland Medical Devices Agency,PMDA) 关于复杂注射剂的注册类型、技术要求及批准的案例,期望为中国的制药企业提供研发思路。
1 、复杂注射剂概述
复杂注射剂具有显著的临床优势,能够达到长效或减毒的目的:例如棕榈酸帕利哌酮注射剂(1个月)(商品名Invega Sustenna)和棕榈酸帕利哌酮注射剂(3个月)(商品名Invega Trinza)可以减少给药次数,增加患者顺应性;将紫杉醇开发成脂质体(商品名力扑素),解除了紫杉醇注射剂中的聚氧乙烯(35)蓖麻油引起的超敏风险,明显降低了紫杉醇注射剂的不良反应;将盐酸多柔比星开发成脂质体,可有效降低心脏毒性[3]。
复杂注射剂技术难度大、壁垒高,不易被仿制,可以长期垄断市场。以美国为例,自首个注射用微球获批上市以来共有10个产品获批上市,至今尚无仿制药获批。
国际上一些制剂的发展经历了从普通口服制剂到复杂注射剂的历程,通过剂型的改良,解决了临床需求,也延长了产品的市场生命周期,例如利培酮(表1)、阿立哌唑(表2)等。


复杂注射剂的市场效应不低于新化学实体新药,表3为几个代表性产品在2016、2017和2018年的全球销售额。
2、欧美日复杂注射剂改良型新药的开发
2.1 复杂注射剂改良型新药在美欧日的注册分类和申报途径
由于各国立法基础不一样,因此适用于复杂注射剂新药上市申请的类型也不尽相同。下文将逐一进行阐述。逐一进行阐述。
2.1.1 复杂注射剂在美国的注册途径
按照FDA相关指南[4],根据新药申请(new drug application,NDA)是否基于申请人自己的研究数据,复杂注射剂改良型新药在美国的注册途径可以是505(b)(1),也可以是505(b)(2)。505(b)(1)申请指的是支持NDA的数据来自于申请人自己开展的研究或者获得使用许可的申请,通常适用于第1类新分子实体的NDA,也适用于在上市产品的基础上进行改变的申请(不包括仿制药),如强生公司的注射用利培酮微球(Risperdal Consta)是在已上市产品利培酮口服制剂的基础上进行的改良、大冢制药的阿立哌唑长效注射剂(Abilify Maintena Kit)是在已上市口服制剂阿立哌唑片(Abilify)的基础上进行的改良,由于都是基于申请人自己研究获得的数据,因此其注册途径为505(b)(1)。505(b)(2)申请指支持NDA的数据不是全部由申请人自行研究获得,而是一部分引用了公开发表的文献或者FDA已批准新药的NDA中的研究内容,并且未获得引用许可的申请,这种申请是为了鼓励药品研发创新,免去重复性研究,并且反映出与仿制药505(j)申请相同的原则:开展研究证明已知的信息既浪费资源又没有必要。适用于505(b)(2)申请的情况有2类:第一类是新分子实体,如Alkermes公司的月桂酰阿立哌唑长效注射剂(Aristada),是在大冢制药的阿立哌唑片(Abilify)的基础上对化合物和制剂进行的改良,其活性成分月桂酰阿立哌唑是由阿立哌唑改变结构变而得,化学分类属于第1类新分子实体,在其NDA中使用了阿立哌唑片(Abilify)的公开数据,但未获得许可,因此属于505(b)(2)申请;第二类指的是与已批准药品的活性成分、剂型、规格、给药途径、处方组成、给药方案和(或)适应证等不同的新制剂,如Abraxis公司的注射用紫杉醇(白蛋白结合型)(Abraxane),使用了HQSPCLT公司的紫杉醇注射液(Taxol)的公开发表信息和FDA的审评审批结论。
2.1.2 复杂注射剂在欧洲的注册途径
与美国505(b)(2)申请类似,欧盟基于2001/83/EC法令相关条款规定了上市许可申请(marketing authorisation applications,MAA)的分类,包括完整的申请(full applications)和仿制药申请(generic applications)、仿改药申请(hybrid applications) 或生物类似药申请(similar biological applications)。完整的申请即符合2001/83/EC法令中第8(3)款内容的申请,按照2001/83/EC法令中第8(3)款内容提交的完整申请必须包括药学(理化、生物或微生物)、临床前(药理毒理)和临床研究的结果。仿改药申请指的是根据2001/83/EC法令中第10(3)款提交的上市申请,这类申请一部分基于参比制剂的临床前和临床研究结果,另一部分基于申请人自行开展试验获得的新数据。与仿制药不同,以下情况需要提交非临床和临床研究结果:①不能满足仿制药的严格定义;②生物利用度研究不能证明生物等效;③活性成分、适应证、规格、剂型或给药途径与参比制剂相比有所改变。例如2018年11月20日按照集中程序在欧盟批准上市的丁丙诺啡长效注射剂(商品名Buvidal),该制剂开展了一系列与Indivior公司的丁丙诺啡舌下片(商品名Subutex)进行对比的临床研究,包括不同规格单次给药和多次给药的药代动力学(PK)对比,参与的受试者包括健康志愿者和患者,建立了群体PK模型,同时还开展了1项与丁丙诺啡舌下片进行对比的非劣效临床研究,用于支持Buvidal的上市申请[5],由于该申请一部分基于参比制剂丁丙诺啡舌下片的试验结果,因此属于仿改药申请。
2.1.3 复杂注射剂在日本的注册途径
日本PMDA没有类似于美欧的从支持上市许可的数据来源进行分类的注册途径,而只是在《日本药事管理法规》中根据活性成分及制剂的情况分为了10类,其中适用于复杂注射剂新药的分类为1类新药-新分子实体、2类新药-新复方制剂:3类新药-给药途径、4类新药-新适应证、5类新药-新剂型、6类新药-新剂量。根据各个产品的PMDA审评报告可知其注册分类,例如棕榈酸帕利哌酮长效注射剂(1个月)属于1类新药-新分子实体;注射用利培酮微球(RisperdalConsta)属于3类新药-给药途径;注射用艾塞那肽微球(Bydureon)同时属于4类新药-新适应证、5类新药-新剂型和6类新药-新剂量。
2.2 复杂注射剂改良型新药的非临床研究要求及案例分析
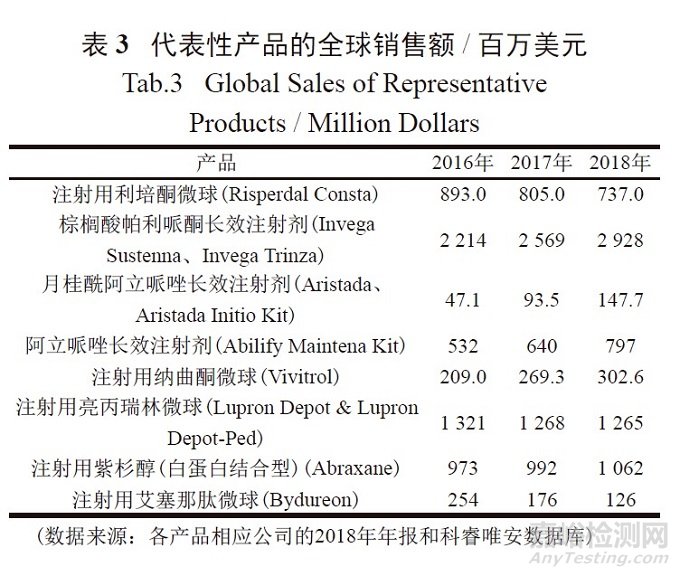
一般情况下,与被改良产品(参比制剂)相比,改良型新药会存在一些不同,比如系统暴露量不同、活性成分不同(比如改盐)、剂型和(或)给药途径不同、处方组成和(或)规格不同、适应证不同、给药剂量不同、属于组合产品等,针对具体情况,需要开展或者能够简化的非临床研究有所不同[6]。需要考虑多种因素来确定需要开展的非临床研究以及试验的数量和类型,主要集中在以下4个方面:①例如,当参比制剂上市很久,当时开展的非临床研究可能无法满足ICH M3指南即《关于实施药物人体临床研究以及上市批准非临床安全性研究的指导原则》的要求[7],在这种情况下,开发505(b)(2)新药可能需要做一些额外的非临床研究以弥补参比制剂研究的不足;②开发的505(b)(2)新药和参比制剂之间的任何不同所产生的安全性都应当进行充分的论证,例如新的给药途径可能需要开展局部安全性试验;③需要论证辅料的安全性;④需要对杂质和降解产物进行界定。对于新制剂和(或)新给药途径的新药,FDA发布了非临床研究指南,用于指导这类产品的非临床研究[8]。与其他改良型新药的非临床研究相似,复杂注射剂非临床研究需要结合具体产品的特点,参照ICHM3 指南合理设计非临床研究策略。从FDA批准的各个复杂注射剂新药的非临床审评报告可以看出,与新分子实体新药相比,相应产品开展的非临床研究的内容有所简化,举例见表4。
2.3 美欧日对复杂注射剂改良型新药的临床研究要求及案例分析
2.3.1 美国FDA的临床研究要求
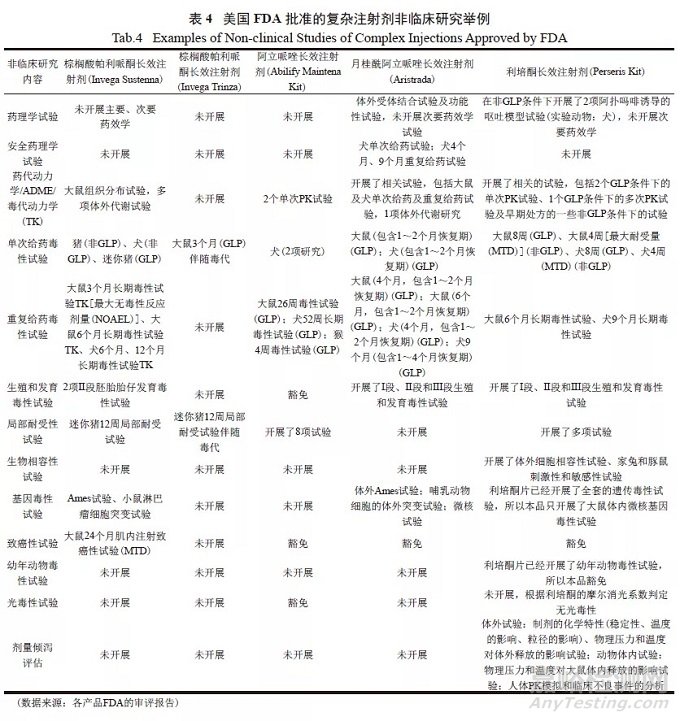
总体来说,FDA对于505(b)(2)新药的临床研究要求遵循个案分析和递进式的原则。除了适用于505(b)(2)的新化学实体新药,505(b)(2)新药基于公开发表的文献和(或)FDA已批准上市药品的安全性和有效性的信息寻求上市批准,因此对于这类新药来说,开展与已批准上市药品的桥接试验是最重要的。505(b)(2)申请最重要的一部分就是开展受试药物与已上市药品(参比制剂)的生物利用度或生物等效性(BE)试验,以比较二者的生物利用度(系统暴露),如果合理的桥接试验能够提供足够的支持,则不需要完整的安全性和有效性研究。如果仅做桥接试验不足以支持,则需要开展其他试验,如食物影响试验、药物相互作用试验、对肝肾等器官损伤的影响试验、合适剂量范围评估试验或纳入群体药代动力学分析的Ⅲ期临床研究,以揭示年龄、性别、体质量等变化对试验药物系统生物利用度的影响[9]。
以FDA在2013年2月28日批准的阿立哌唑长效注射剂(商品名为Abilify Maintena Kit) 为例,为支持NDA,申请人开展了3个临床药理学研究:单次给药体内释放特性试验(CN138-020,开放、两阶段、非随机试验,精神分裂症或者精神分裂障碍患者)、单次给药PK试验(31-007-02)、多次给药PK试验(31-07-244,开放、多次给药,精神分裂症患者),申请人开展了群体PK分析和模拟以评估药物-药物相互作用和药物突释。由于阿立哌唑已被批准用于治疗精神分裂症,FDA要求申请人开展1项评估注射剂有效性的研究(31-07-246,52周、多中心、随机、双盲、安慰剂对照的临床研究,精神分裂症患者),注射剂的安全性则基于3个PK试验和7个Ⅲ期研究,包括在美国开展的安慰剂对照试验(31-07-246)、在欧洲开展2个阳性药对照试验(31-07-247和31-08-003)以及4个大型的开放性安全性试验(31-08-248、31-10-270、31-10-002和31-11-283),安全性数据来自共计1324例至少接受1次给药的患者和1287例接受1次或多次每月肌内注射300或400mg制剂的患者,其中832例患者至少接受了6个月的给药、630例患者至少接受了12个月给药[10]。
以FDA在2015年10月5日批准的月桂酰阿立哌唑长效注射剂(商品名Aristada)为例,该药共有4个规格,441、662、882和1064mg,用于用于治疗成人精神分裂症。该药的注册分类虽然为第1类新分子实体,但是由于依赖于FDA批准的参比制剂阿立哌唑片(商品名Abilify)的安全性和有效性的信息,因此属于505(b)(2)申请。申请人为建立Aristada与参比制剂Abilify之间的PK桥接,开展了4个Ⅰ期临床研究(ALK9072-001、ALK9072-101、ALK9072-002、ALK9072-102)和1个Ⅲ期临床研究(ALK9072-003),建立了群体PK模型,通过拟合从2个Ⅰ期临床研究(ALK9072-001、ALK9072-002)和1个Ⅲ期临床研究(ALK9072-003)中获得的阿立哌唑片的数据和从5个试验汇总获得的肌内注射月桂酰阿立哌唑的数据,获得肌内注射月桂酰阿立哌唑相对于参比制剂阿立哌唑片的相对生物利用度为58%。Alkermes公司在NDA中提交的数据可以说明,在批准的阿立哌唑片的剂量范围内阿立哌唑的稳态暴露量为每天10~30mg,肌内注射月桂酰阿立哌唑后阿立哌唑的暴露量与该水平可比,这一可比性为月桂酰阿立哌唑推荐的剂量和给药方法的有效性提供了证据[11]。可以看出,尽管是新分子实体新药,但基于与已上市参比制剂的桥接及已上市参比制剂的安全性和有效性数据,支持上市申请的临床研究也比常规的、按照505(b)(1)申请上市的新分子实体药物的临床研究要求大大简化。
由于Aristada给药后需要3周才能达到稳态血药浓度水平,因此首次注射需要连续21d口服阿立哌唑片以达到有效治疗浓度,对于精神分裂患者来说,21d口服给药的顺应性是个很大的挑战,因此Alkermes公司开发了另1款月桂酰阿立哌唑注射混悬剂,该制剂于2018年6月29日被FDA批准上市(商品为AristadaInitio Kit),规格为675mg∶2.4ml,与口服阿立哌唑联合作为Aristada治疗成人精神分裂症的启动方案,首次给药需要注射Aristada并同时注射AristadaInitio Kit 675mg及口服阿立哌唑30mg。该药只开展了3项PK试验,分别为随机、开放、安慰剂对照的单次给药剂量递增试验;随机、双盲、阳性药对照的试验,以比较Aristada的2个启动方案的PK、安全性及耐受性(关键性PK桥接试验);以及随机开放的单次给药试验,未开展其他的安全性和有效性临床研究。关键性PK桥接试验的结果说明采用Aristada Initio Kit 启动的方案与使用Aristada同时联合服用21d阿立哌唑片的预期暴露量相似[12]。
以2018年7月27日FDA批准上市的利培酮1个月制剂(PerserisKit)为例,该产品是在利培酮已上市制剂的基础上开发的1种新的复杂注射剂,规格为90和120mg,PerserisKit 由2个预灌装注射器组成,1个注射器里装的是利培酮粉末,另1个注射器装的是由聚丙交酯-乙交酯聚合物和N-甲基-2-吡咯烷酮组成的递药系统,使用前将二者混合形成混悬液,皮下注射后能使Perseris Kit中的利培酮释放1个月。该产品按照505(b)(2)途径申报,参比制剂为口服利培酮片,申请上市时开展了2项Ⅰ期临床研究:单次给药试验(RB-US-09-0007)和单次给药剂量递增试验(RB-US-09-0008),1项Ⅱ期开放的多次给药剂量递增试验(关键的PK桥接试验,RB-US-09-0009),1项随机、安慰剂对照的有效性和安全性的Ⅲ期临床研究(RB-US-09-0010),以及1项开放的长期安全性的Ⅲ期临床研究(RBUS-13-0005),其中关键PK桥接试验证明了皮下注射Perseris Kit 60~120mg的药代动力学数据呈线性,并在第2个月剂量结束时达到稳定水平,皮下注射在稳态下的波动较小,Perseris Kit的平均总利培酮浓度与口服利培酮片的水平相当[13]。
2.3.2 EMA的临床研究要求
EMA发布的《治疗精神分裂症的包括储库式制剂在内的药物研究指南》中提到了治疗精神分裂症的储库式制剂的临床开发策略[14],主要包括:①通过PK研究来确定新制剂属于储库式制剂;②比较储库式制剂与口服制剂的生物利用度以评价活性成分可接受水平的持续时间;③在病情稳定的患者身上比较储库式制剂与口服制剂的有效性;④解决从口服制剂转换到储库式制剂的问题。原则上,除非证明储库式制剂与口服制剂具有明确的药代动力学或药效学关系,否则储库式制剂必须进行临床研究,以比较口服制剂和储库式制剂的有效性并证明剂量间隔的合理性。据此可以看出EMA对于仿改药申请的临床研究的要求也是渐进式和个案分析。针对同一产品,欧美对相关临床研究的要求有可能不同,以阿立哌唑长效注射剂(商品名AbilifyMaintena Kit) 为例,从EMA发布的审评概述中可以看出:申请人开展的安慰剂对照的试验31-07-246未包括与口服阿立哌唑的直接对比,且采用了与阳性药对照的试验31-07-247不同的主要有效性终点,EMA认为试验31-07-247才是关键性试验,而试验31-07-246是支持性的临床研究[15],这点与FDA不同。
2.3.3 日本PMDA的临床研究要求
以阿立哌唑长效注射剂(商品名Abilify Maintena Kit) 为例,该产品2014年1月15日在日本提交上市申请、2015年2月20日获批上市,支持日本上市申请的临床研究,除了来自海外的3项Ⅰ期临床研究(CN138-020、31-05-244和31-11-289)和2项Ⅲ期临床研究(31-07-246和31-07-247)之外,还有2项在日本开展的Ⅰ期临床研究和1项纳入日本受试者的国际多中心的阳性药对照的Ⅲ期临床研究(长效注射剂组228例,其中日本人118例;阿立哌唑片组227例,其中日本人119例)[16]。
3 、美日欧对复杂注射剂仿制药的技术要求及案例分析
一般情况下FDA要求仿制药与参比制剂相比具有相同的化学成分、适应证、给药途径、用法用量,并不要求辅料相同。对于注射剂,根据FDA《联邦管理法》314.94章的第(9)(iii)条要求,仿制药所用的辅料的种类和用量与参比制剂相同(Q1Q2),抑菌剂、缓冲剂(含常规的pH调节剂)或抗氧剂可以不同,但要有充分的依据。辅料的用量相同是指仿制药辅料用量为参比制剂相应辅料用量的95%~105%。如果达不到这个条件,仿制药的简化新药申请(abbreviated new drug application,ANDA) 会被FDA拒绝接受[17]。FDA为指导企业进行复杂注射剂的研发,制定了许多相应的举措,例如针对具体品种发布了相应的仿制药研究指南[18],规定了BE试验的基本要求,如试验个数、试验设计、受试者、规格、多规格可豁免某些规格的要求,以及体外释放度试验的要求,少部分品种甚至给出了产品质量要求,如盐酸多柔比星脂质体。此外,FDA重视与申请人的沟通交流,一方面申请人可以书面咨询FDA[19],另一方面申请人可以申请与FDA召开沟通交流会[20],讨论具体品种的问题。
与FDA不同,EMA并未要求注射剂仿制药要满足Q1Q2。EMA也会针对具体品种发布仿制药研究指南[21],但总体数量比FDA少且仅是针对人体BE试验的要求,无体外试验的相关要求。目前EMA发布了注射用艾塞那肽微球、注射用醋酸奥曲肽微球、棕榈酸帕利哌酮长效注射剂、盐酸多柔比星脂质体等复杂注射剂的BE指南。FDA和EMA针对同一品种的BE要求也会存在差异,详见表5。
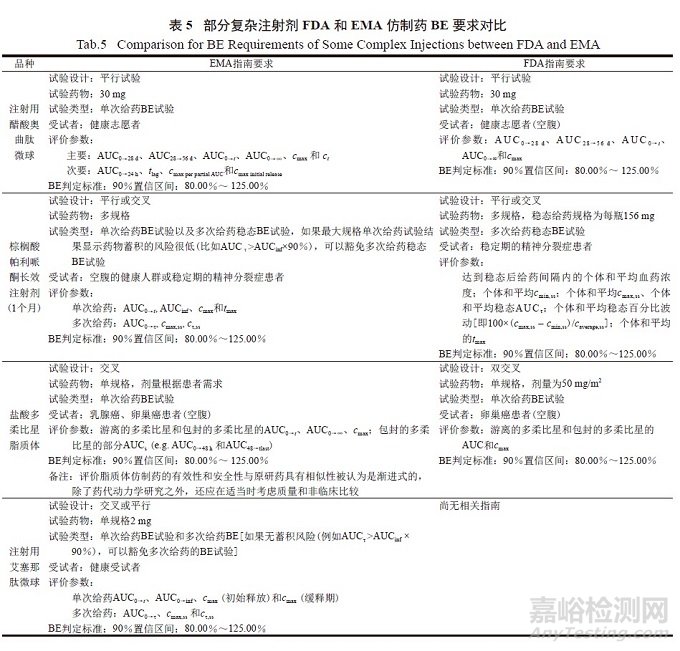
日本PMDA未针对具体品种发布仿制药研究或者BE试验指南,一般情况下参照《仿制药生物等效性试验指导原则》[22]。从日本批准的亮丙瑞林微球仿制药的BE试验情况可以看出[23],与FDA发布的亮丙瑞林微球(1月制剂)仿制药指南中的BE要求并不完全相同。FDA指南要求在接收初始治疗的前列腺癌患者中开展随机、平行、单次给药的BE试验,采用7.5mg开展BE试验,其他规格(3.75、11.25和15mg)满足条件可豁免,测定血浆中亮丙瑞林,评价参数为AUC7→t、AUC0→t、AUC0→∞和cmax;日本批准的亮丙瑞林微球仿制药则是2个规格(1.88和3.75mg)分别与原研药2个规格(1.88和3.75mg)开展了单次给药的BE试验,受试者为24例绝经后的健康成年女性,测定血浆中亮丙瑞林药物浓度,计算药代动力学参数cmax、AUC0→42d、AUC0→7d、AUC7→28d,结果达到生物等效[23]。
4、 结语
复杂注射剂因具有独特的制剂特点,可以满足未被满足的临床需求,一直是国内外新制剂开发的热点之一,但其开发难度较大,一方面是药学的研发,需要有相关制剂的技术平台、适合的辅料和商业化生产的能力,同时,在保持处方组成与参比制剂一致的前提下,很难突破原研药的专利;另一方面是临床开发策略,需要根据制剂自身的特点合理设计临床研究,加快上市步伐。从美欧日批准上市的产品来看,各国药监部门对于这类产品尤其是改良型新药的非临床和临床研究的要求基本都是基于个案分析和渐进式的原则。因此,如何与已上市参比制剂(通常是原研药)进行药代动力学桥接,充分利用已上市产品的安全性有效性数据,进而减免不必要的非临床和临床研究、提高试验成功率,对申请人来说是技术和策略制定的难点,对监管机构来说也是审评难点,需要双方充分沟通交流,以便就这些产品的非临床和临床研究要求达成共识,促进其尽早上市,惠及患者。
对于复杂注射剂,尽管FDA和EMA都针对具体品种制定了研究指南,规定了BE试验的要求,但这些指南是基于美国和欧盟的监管和临床实践,我国临床实践不同,例如,FDA发布的棕榈酸帕利哌酮长效注射剂(1个月)仿制药研究指南中要求BE试验的受试者为已经接受帕利派酮缓释混悬液肌内注射给药方案的稳定的男性和非妊娠女性精神分裂症或分裂情感性障碍患者,实践中由于该产品在美国上市较早,有一定的临床使用基础,而我国进口上市时间较晚,且价格昂贵,临床使用非常有限,因此满足条件的受试者非常少,BE试验开展起来受试者入组难度相当大。我国应当从科学、伦理、用药可及性和可支付性出发,制定适用于我国的仿制药研究指南,以促进这类高难度仿制药的研发和早日上市。

来源:中国医药工业


