您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-09-24 14:03
2020年9月16日,FDA在发布物理医学器械;非侵入性骨生长刺激器的重新分类的通报,主要内容包括:
1)提议将非侵入性骨生长刺激器(产品代码LOF和LPQ)从III类(上市前批准)重新分类为II类(特殊控制),并将遵守上市前通知(510(k))的要求。
2)提出了名称为“非侵入性骨生长刺激器”的新器械分类以及FDA认为有必要对这些器械提供合理安全性和有效性保证的特殊控制。
3)可在2020年10月16日之前提交相关电子或书面评议意见。
1.产品描述
非侵入性骨生长刺激器目前指定为产品代码LOF和LPQ,通常由波形发生器和换能器(例如,线圈、电极和/或超声换能器)组成。患者接触表面包括传感器、电极导线和器械外壳。非侵入性骨生长刺激器利用电气组件来产生通过无创应用传感器(例如,电磁线圈或超声传感器)或电极(例如,电容器板)输送到治疗部位的输出电、磁或超声波形。该器械还包括监测输出波形和治疗输送的内部方法,并提供视觉和/或听觉报警,以警告用户器械功能不当。
非侵入性骨生长刺激器通常用于促进成骨,作为骨折固定或脊柱融合主要治疗的辅助治疗。该器械类型的适应症取决于具体的器械特征,但包括:治疗继发于阑尾系统创伤的已确定骨不连、治疗先天性假关节、治疗阑尾系统融合失败、某些新鲜骨折的早期治疗,并作为腰椎或颈椎融合的辅助治疗。
2.重新分类的原因
根据FD&C法案第513(f)(3)部分和21 CFR第860部分C子部分,FDA提议根据产品代码LOF和LPQ将非侵入性骨生长刺激器器械从III类重新分类为II类,这包括使用CC、PEMF和CMF以及超声信号产生电场或磁场的器械。FDA认为,通过FDA从上市前申请、同行评审文献、医疗器械报告(MDR)的评审中积累的这些器械的经验,可以获得足够的信息对确定的风险建立特殊控制,有效缓解确定的健康风险,以提供合理的安全性和有效性保证。
《FD&C法案》第510(m)条规定,如果FDA确定无需上市前通告来合理保证器械的安全性和有效性,则可以豁免第II类器械的《FD&C法案》第510(k)条规定的上市前通告要求。对于非侵入式骨生长刺激器,FDA已确定需要上市前通知来合理保证器械的安全性和有效性。因此,FDA不建议豁免这些拟定的II类器械的510(k)要求。
具体来说,FDA建议要求将临床性能数据作为特殊控制来解决成骨失败或延迟的风险。FDA的提案要求对任何非侵入性骨生长刺激器的临床性能进行评价,以支持其预期用途。
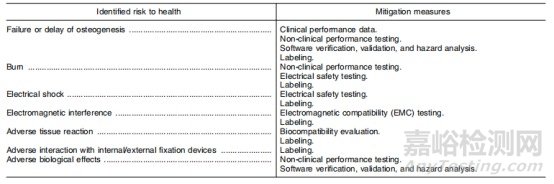
3.健康风险及特殊控制
FDA要求提交非侵入性骨生长刺激器510(k)的公司应遵守特殊控制中规定的特定缓解措施。除一般控制外,必须遵守特殊控制,以合理保证这些器械的安全性和有效性。
非侵入性骨生长刺激器的健康风险和缓解措施如下表所示:

来源:医课汇


