您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-08-04 19:54
|
NDA序号 |
217900 |
|
申请人 |
Sun Pharmaceutical Industries, Inc |
|
药物名 |
LEQSELVI (deuruxolitinib), 片剂8 mg |
|
剂型 |
片剂 |
|
拟商业化规格 |
8 mg |
|
使用途径 |
口服 |
|
每日最大剂量 |
16mg |
|
Rx/OTC |
Rx |
|
适应症 |
用于治疗成人中度至重度脱发 |
溶解度:
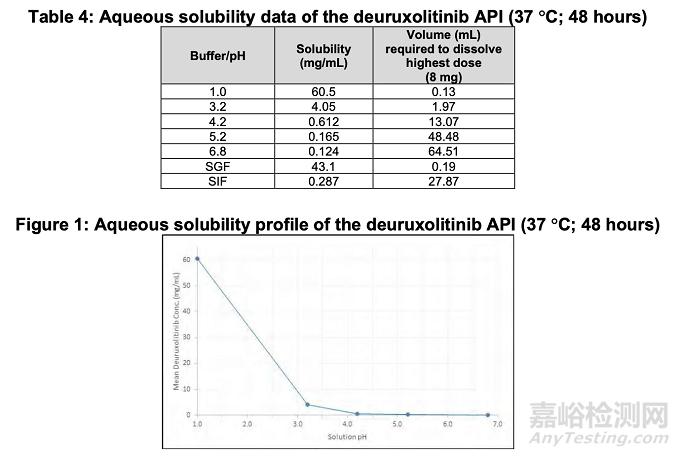
活性药物成分 (API) deuruxolitinib 为白色至类白色结晶固体,在低 pH 条件下具有高水溶性,在多种有机溶剂中呈现良好至差的溶解度。Deuruxolitinib 原料药在生理 pH 范围内的水溶性曲线(如下表 4 和图 1 所示)显示其溶解度呈 pH 依赖性变化,范围从 60.5 mg/mL(pH 1.0)至 0.124 mg/mL(pH 6.8)。
渗透性:
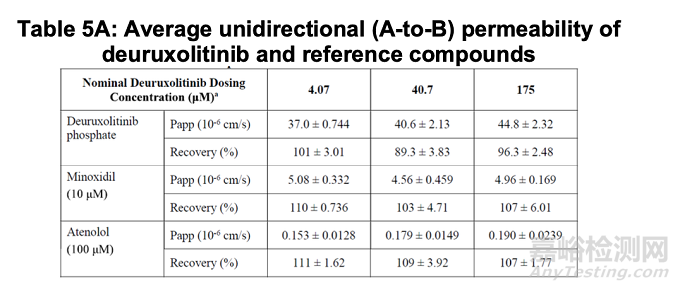
在Caco-2细胞单层测试系统中,deuruxolitinib原料药在4.07 µM(32 mg剂量于250 mL中的1% [407 µM游离碱])、40.7 µM(10%)及175 µM(43%)浓度下的顶侧-基底外侧表观渗透性(Papp)均高于高渗透性参比化合物米诺地尔(表5A)。因Caco-2细胞测试系统的耐受性问题,无法使用407 µM(100%)浓度进行实验。
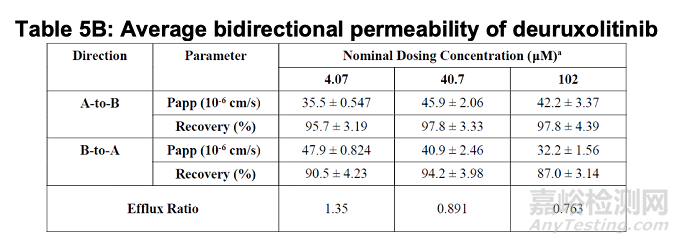
评估双向(从A到B和从B到A)渗透性的三种deuruxolitinib给药浓度分别为:4.07 µM(相当于32 mg剂量溶于250 mL溶液时游离碱浓度407 µM的1%)、40.7 µM(10%)及102 µM(25%)。在测试浓度范围内,外排比率介于1.35至0.763之间(见表5B)。
除了Caco-2细胞渗透性研究外,还通过向健康志愿者给予放射性标记的deuruxolitinib进行了一项人体吸收和代谢研究(研究编号CP543.1004)。在健康成年男性受试者中单次口服deuruxolitinib后,平均90%的给药放射性被回收,其中约70%在尿液中回收(主要为代谢物),约20%在粪便中回收(主要为代谢物)。原型deuruxolitinib占排泄物中放射性的比例不足2%。
FDA评估:
根据deuruxolitinib原料药在生理pH值1.0至6.8范围内的溶解度特性,其最高剂量强度8mg在最低溶解度0.124mg/mL时(pH 6.8条件下,见表4),溶解所需溶剂体积为64.51mL。需说明的是,溶出度开发和体内药代动力学研究均基于12mg规格制剂开展。对于12mg剂量强度及0.124mg/mL的最低溶解度,溶解所需溶剂体积为96.77mL。因此,deuruxolitinib原料药可视为高溶解性化合物。基于Caco-2渗透性及人体吸收代谢研究结果,该原料药亦可视为高渗透性化合物。
根据BCS分类体系,申请人将德鲁昔替尼原料药归为BCS 1类化合物。但申请人未提出正式的BCS分类认定请求,故该原料药的渗透性及BCS分类尚未经评估认定。
溶出方法与接受标准
B.2.1. 体外溶出度测定法:
在制剂开发的早期阶段,基于原料药溶解度和剂型的速释特性,确定了溶出介质及体积。作为溶出方法开发的一部分,对以下溶出参数进行了研究:
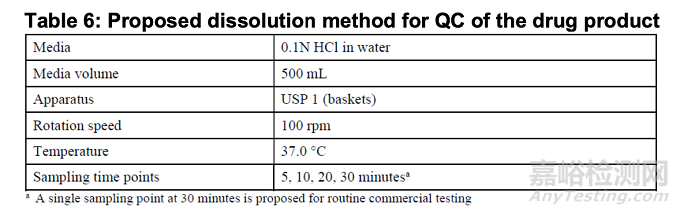
具体细节详见溶出度方法开发报告。申请人用于产品放行时和在稳定性研究期间的溶出度质量控制方法,遵循2018年8月发布的FDA行业指南《含高溶解度原料药的速释固体口服制剂溶出度测试及验收标准》中的方法,详见表6:
FDA评估:
根据deuruxolitinib原料药在生理pH值1.0至6.8范围内的溶解度特性,在最高剂量强度8毫克和最低溶解度0.124毫克/毫升(pH 6.8;表4)条件下,溶解该原料药所需溶剂体积为64.51毫升。需注意,溶出度开发及体内药代动力学研究基于12毫克剂量规格药品进行。对于12毫克剂量强度和0.124毫克/毫升最低溶解度,溶解该原料药所需溶剂体积为96.77毫升。
因此,deuruxolitinib原料药可视为高溶解度化合物。12毫克剂量产品单次给药I期研究(研究编号CP543.1009)显示中位达峰时间(Tmax)为0.5-0.88小时,符合该剂型的即速释特性。故所提接受标准符合美国FDA 2018年8月颁布的《含高溶解度原料药的速释固体口服制剂溶出度测试及接受标准》工业指南方法,可予认可。鉴于deuruxolitinib原料药在pH 1.0-6.8生理范围内呈现高溶解度,且溶出方法符合指南要求,从生物药剂学角度风险极低,无需证明该方法的区分能力,亦无此必要。
B.2.2. 溶出度数据与接受标准:
根据溶出方法,申请人提出的用于产品放行及稳定性研究期间质量控制的接受标准为"30分钟时Q值=X"。申请人申报的用于药物产品质量控制的溶出方法及接受标准详见表7。

FDA评估:
本品为速释制剂(达峰时间中位数为0.5−0.88小时),所含原料药具有高溶解性。(b)(4)根据全部提交信息,认为表7所示溶出度测定方法及接受标准符合要求。8mg与12mg规格临床批次及拟上市批次的溶出数据链接详见附录1。
B.2.3. 稳定性研究中的溶出数据:
下表8所示为拟采用的溶出度方法/可接受标准,对四个8mg规格注册批次进行的稳定性研究结果。原始稳定性数据在长期(25°C/60% RH)、加速(40°C/75% RH)及中间储存条件(30°C/65% RH)下生成。
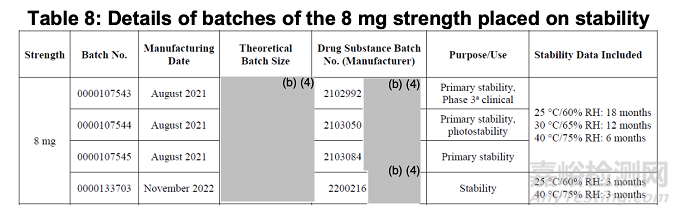
申请人尚未进行体内外相关性(IVIVR/IVIVC)或任何模型研究。其提出的溶出度方法旨在确保批间一致性,此方案可接受。
基于该产品12 mg规格的1期药代动力学研究(研究编号CP543.1009),该产品中位达峰时间(Tmax)范围为0.5至0.88小时。速释制剂较短的Tmax得到溶出数据支持——在生理相关溶出条件下,30分钟时间点产品溶出度>90%。
药品快速溶出特性符合"30分钟内溶出度Q=%"的验收标准。由于deuruxolitinib原料药在速释剂型中属高溶解度原料药,从生物药剂学角度评估风险极低,故区分能力的证明为非关键要素。
【学习笔记】
从原料药的溶解度数据来看,化合物呈现pH值依赖性溶解,pH值越高,溶解度越低。但由于药品的拟上市规格并不高,为8mg,因此在常见的溶出体积500ml、900ml中都有很高的溶解度,因此8mg在高低pH值的介质中都具有高溶解性。
渗透性研究表明,化合物具有高渗透性,结合其溶解度的数据,化合物具有高溶解高渗透行为,表现为BCS I类化合物性质。
溶出方法的选择是基于化合物的类BCS I类的性质,参考FDA指南,高溶解性化合物得到的速释口服固体制剂的溶出测试指南,选择了指南中推荐的溶出方法,而且可以不再考察方法的区分力;同时根据高规格12mg的体内中位达峰时间,判断该高溶解性化合物的口服固体速释制剂,生物药剂学风险非常低。
溶出标准的设定参考了关键批次,注册批次的放行数据和稳定性数据,皆表现为快速溶出,同时也参考了12mg规格的中位达峰时间,完全符合速释制剂特征,30分钟内体外的溶出数据能达到90%以上。基于以上数据和分析,溶出方法的区分力特性不是关键因素,接受限度也就易制定。

来源:蒲公英Ouryao


