枸橼酸西地那非是一种环磷酸鸟苷(cGMP)特异的 5 型磷酸二酯酶(PDE5)的选择性抑制剂 ,PDE5广泛存在于血管平滑肌、呼吸道平滑肌、血小板、胃肠道上皮细胞等,通过特异性水解细胞内第二信使 cGMP,参与多种生理、病理过程的调控,是良好的药理学靶标。枸橼酸西地那非是选择性PDE5 抑制剂,通过抑制血管平滑肌上的 PDE5,提高局部 cGMP 水平,松弛、扩张血管平滑肌,广泛用于勃起功能障碍和肺动脉高压(PAH)的治疗。
查询美国食品药品监督管理局(FDA)及欧盟药品监督管理局(EMA)网站相关信息,枸橼酸西地那非片由辉瑞公司研发,1998 年 3 月在美国作为新分子实体(Type I)获得批准[新药申请(NDA)号020895],商品名为 Viagra®,规格 25、50、100 mg,成为美国批准的第1个用于治疗男性勃起功能障碍的口服治疗药物;2005年6月,辉瑞公司的枸橼酸西地那非片在美国被批准用于治疗肺动脉高压(NDA号 021845),商品名 Revatio®,规格 20 mg;1998 年获欧盟批准上市;目前该产品已在美国、英国、日本以及欧盟多个国家或地区批准上市。辉瑞公司生产的原研地产化枸橼酸西地那非片于 2000 年 5月在中国获批上市,商品名万艾可®,涉及 25、50、100 mg 规格,2020 年 2 月该公司 20 mg 规格枸橼酸西地那非片获批进口中国。
截至 2022 年 8 月 31 日,中国已批准 30 个枸橼酸西地那非片(含口崩片)仿制药批准文号(含 2 个境外进口、3个原研地产化产品批准文号)、1个原研进口批准文号,涉及该品种的 4 个规格(20、25、50、100 mg);经查国家食品药品监督管理局(NMPA)药品审评中心化学药品目录集(https://www.cde.org.cn/hymlj/listpage/9cd8db3b7530c6fa0c86485e563f93c7),共收载 26 条枸橼酸西地那非片(含口崩片)信息,含原研地产化产品3条(标记为参比制剂),23个国内批准文号通过或视同通过化学仿制药一致性评价。经查询药物临床试验登记与信息公示平台(http://www.chinadrugtrials.org.cn/index.html),截至 2022 年 8 月 31 日,共登记枸橼酸西地那非片(含口崩片)生物等效试验 50条,其中 31条登记状态为已完成、18条为进行中、1条为主动终止。
通过梳理国内、外药品监管机构对枸橼酸西地那非片生物等效性试验的要求,结合近年来该药在中国的生物等效性试验现状,分析该品种生物等效性试验的主要特征,结合生物等效性试验审评过程中遇到的多种情况提出考虑,以期为提高仿制药质量有所裨益。
一、体内药动学特征
枸橼酸西地那非片口服后吸收迅速,绝对生物利用度为 41%(25%~63%)。其药动学参数在推荐剂量范围内与剂量成比例。消除以肝脏代谢为主[细胞色素P450同功酶3A4(CYP4503A4)途径],生成有活性的代谢产物 ,其性质与西地那非近似。CYP4503A4的强效抑制剂(如红霉素、酮康唑、伊曲康唑)以及 CYP450 的非特异性抑制物如西咪替丁与西地那非合用时,可能会导致西地那非血浆水平升高。西地那非及其代谢产物的消除半衰期约为4 h。
1.1 吸收和分布
枸橼酸西地那非片吸收迅速。空腹状态下口服 30~120 min(中位值 60 min)后达到最大血药浓度(Cmax)。在与高脂肪饮食同服时,吸收速率降低,达峰时间(tmax)平均延迟60 min,Cmax平均下降29%。但吸收程度未受显著影响[药时曲线下面积(AUC)下降 11%]。西地那非浓度为 3.5 nmol·L−1时对人PDE5 酶活性的体外抑制率达 50%。在人体中,单剂口服100 mg西地那非后,平均最大游离血浆西地那非浓度约为 18 ng·mL−1或 38 nmol·L−1。西地那非的平均稳态分布容积(Vss)为 105 L,说明其在组织中有分布。西地那非及其主要循环代谢产物(N-去甲基化物)均有大约96%与血浆蛋白结合。蛋白结合率与药物总浓度无关。
1.2 代谢和排泄
西地那非主要通过肝脏的微粒体酶细胞色素P4503A4(主要途径)和细胞色素 P4502C9(次要途径)清除。主要循环代谢产物是西地那非的N-去甲基化物,后者将被进一步的代谢。N-去甲基代谢产物具有与西地那非相似的磷酸二酯酶(PDE)选择性,在体外其对 PDE5 的作用强度约为西地那非的50%。此代谢产物的血浆浓度约为西地那非的40%,故西地那非的药理作用大约有 20% 来自于其代谢产物。N-去甲基化代谢产物被进一步代谢,其终末半衰期约为4 h。西地那非的全身清除率为41 L·h−1,终末半衰期为 3~5 h。口服或静脉给药后,西地那非主要以代谢产物的形式从粪便中排泄(约为口服剂量的 80%),一小部分从尿中排泄(约为口服剂量的13%)。
二、药品监管机构的生物等效性试验要求
2.1 美国FDA技术要求
美国FDA自1980年发布第1版《具有治疗等效性的已批准药物》(通常被称作橙皮书)以来,每年3月发行,现行版的橙皮书为第 41 版,其中明确指定了用于仿制药药学和生物等效性研究的参比制剂。FDA自2010年6月开始陆续公布和更新《特定药物的生物等效性指导原则》,为中国化学仿制药一致性评价以及仿制药的上市申请中生物等效性研究提供了指导与参考。美国 FDA 橙皮书中 ,辉瑞公司的商品名为Viagra(® NDA 号 N20895)的枸橼酸西地那非片(规格 25、50、100 mg)与 该 公 司 商 品 名 为Revatio(® NDA 号 021845)的枸橼酸西地那非片(规格 20 mg)均 标 记 为 参 比 制 剂(RLD);其 中 20、100 mg 规格同时标记为生物等效性研究对照药品(RS)。美国 FDA 枸橼酸西地那非片生物等效性研究指导原则( 2008年5月定稿,2008年12月修订)中对本品生物等效性研究建议为空腹及餐后双交叉体内研究设计,采用健康男性受试者,考察单次给药后血浆中西地那非及代谢活性产物 N-去甲基西地那非的主要药动学参数 AUC 及 Cmax 90% 置信区间(90%CI)在80.00%~125.00%,具体要求见表1。
2.2 中国NMPA技术要求
中国于 2021 年 12 月发布《枸橼酸西地那非口崩片生物等效性研究指导原则》,与 FDA 枸橼酸西地那非片生物等效性研究个药指南相比,具有以下 4 点异同:(1)研究类型方面,仅要求开展空腹生物等效性试验;(2)给药方法方面,参考枸橼酸西地那非口崩片说明书给药方法,建议采用直接服用方式开展临床试验,即将口腔崩解片置于舌上,待其崩解后直接吞咽,观察并记录口崩片在口中完全崩解的时间及口感等;(3)给药剂量方面,考虑该品种目前仅批准 50 mg 规格,故建议采用 50 mg 单片服用的给药方法,且不涉及其他规格的生物等效性豁免;(4)其他项目基本与 FDA 枸橼酸西地那非片保持一致。国内现已批准1家企业的枸橼酸西地那非口崩片(50 mg规格)。中国已有10余家企业申报的枸橼酸西地那非片(涉及20、25、50、100 mg规格)获批上市,其生物等效性研究主要参考FDA枸橼酸西地那非片生物等效性研究个药指南,结合2016年中国发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》的总体技术要求开展相关研究。
2.3 欧盟及日本生物等效性研究技术要求
欧盟及日本未发布枸橼酸西地那非片生物等效性研究技术要求。EMA主要参照美国 FDA相关要求,开展本品种的生物等效性研究的技术审评,日本药品和医疗器械署(PMDA)则通过受试制剂与参比制剂在不同介质中有分辨力的溶出曲线代替生物等效性研究批准上市。
三、枸橼酸西地那非片在中国的生物等效性试验评价结果分析
参考药审中心近年来枸橼酸西地那非片 10 余家申请人生物等效性研究申报数据、该品种通过仿制药一致性评价信息公开数据(https://www. cde.org. cn/yzxpj/listpage/baccb6ea4350170164a8141548c32f2e)以及参考文献,血浆中的西地那非及 N-去甲基西地那非主要药动学参数 Cmax、AUC、tmax等的90%CI、变异系数(CV)进行汇总,形成枸橼酸西地那非片生物等效性试验结果分析见下文,部分数据见表2、3。
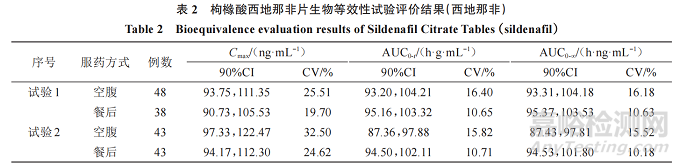
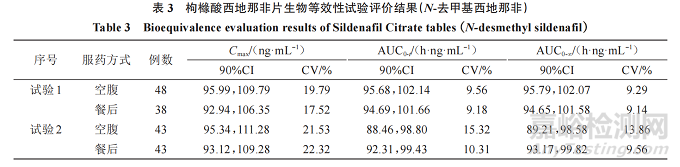
主要药动学参数:生物等效性研究结果显示,西地那非 Cmax最低的 90%CI下限为 80.31%,最高的90%CI 上限为 122.47%;AUC0-t最低的 90%CI 下限为87.36%,最高的90%CI上限为109.92%;AUC0-∞最低的 90%CI 下限为 87.01%,最高的 90%CI 上限为111.57%;N-去甲基西地那非最低的 90%CI 下限为81.01%,最高的 90%CI上限为 116.78%;AUC0-t最低的 90%CI 下 限 为 88.46%,最 高 的 90%CI 上 限 为114.86%;AUC0-∞最低的90%CI下限为89.21%,最高的90%CI上限为114.59%。
CV:生物等效性研究结果显示,西地那非 Cmax的 CV 范围为 18.17%~32.5%、AUC0-t的 CV 范围为10.25%~17.00%、AUC0-∞ 的 CV 范 围 为 10.03%~17.12%;N- 去 甲 基 西 地 那 非 Cmax 的 CV 范 围 为9.78%~27.23%、AUC0-t 的 CV 范 围 为 7.45%~15.32%、AUC0-∞的CV范围为7.77%~15.00%。
四、枸橼酸西地那非片生物等效性试验审评考虑
4.1 参比制剂的选择
截至2022年8月31日,经检索国家药品监督管理局已发布《化学仿制药参比制剂目录》第十批、第十二批、第二十三批、第二十七批、第四十批及第五十五批中发布枸橼酸西地那非片(含口崩片)参比制剂累计11条,涉及20、25、50、100 mg 4个规 格 ,包括辉瑞公司原研进口 、原研地产化产品 、欧盟上市及美国上市的多个持证商的该产品。
参考美国 FDA 枸橼酸西地那非片生物等效性个药指南的相关说明,枸橼酸西地那非片生物等效性研究需选择对应的参比制剂开展生物等效性试验,对于不同的参比制剂需分别提交申请,目前国内大多申请人选择国内已批准的原研地产化产品(持证商为辉瑞制药有限公司、商品名 Viagra®或万艾可®)开展相关研究。由已发布的《化学仿制药参比制剂目录(第二十七批)》、《化学仿制药参比制剂目录(第五十五批)》枸橼酸西地那非片参比制剂发布情况可知,美国橙皮书列为参比制剂的另一个枸橼酸西地那非片原研产品(商品名 Revatio®或瑞万托®),亦有申请人提出申请并开展仿制研究。
4.2 受试者数量、采样点及清洗期设计
已有生物等效性研究数据可见,枸橼酸西地那非片 AUC 及 Cmax的个体内变异为 16%~26%,如果检验的显著性水平设置为 0.05,检出受试制剂与参比制剂生物等效的研究功效为0.9,等效界限设定为 80.00%~125.00%,假设西地那非主要终点的几何均值比为 0.95~1.07,采用 PASS 计算样本量,则估计样本量为 40 例,考虑约 20% 脱落风险,一般空腹及餐后入组男性健康受试者48例左右;采用单剂量、空腹与餐后,两制剂、两周期、两序列、随机、开放、自身交叉的生物等效试验设计。
采样点及清洗期方面,参考中国2016年发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》相关要求,建议每位受试者每个周期采样12~18个采血点,采样时间不短于 3 个末端消除半衰期,末端消除相应至少采集 3~4 个样品以确保准确估算末端消除相斜率,AUC0-t至少覆盖 AUC0-∞的 80%。现有研究结果表明,西地那非及其代谢物N-去甲基西地那非的终末半衰期均为3~5 h,大多研究者选择服药0 h(服药前1 h内)和服药后至24 h的18~20个采血点,清洗期一般不应短于7个半衰期,选择7 d。
4.3 生物等效性评价
由 FDA、我国已发布的枸橼酸西地那非片生物等效性试验个药指南可知,本品种的生物等效性评价要求基本一致,即受试制剂与参比制剂血浆中西地那非主要药动学参数 Cmax、AUC0-t、AUC0-∞的几何均值比值的 90%CI 在 80.00%~125.00%,同时提交其 代 谢 物 N- 去 甲 基 西 地 那 非 的 Cmax、AUC0-t 和AUC0-∞用于进一步支持临床疗效的可比性。
考虑受试制剂与参比制剂在生物等效性研究中药动学参数 tmax能在一定程度上反映 2 个制剂在体内的达峰时间,一般要求申请人在提交统计分析结果是同时提交tmax非参数检验结果,从而进一步保障受试制剂与参比制剂的临床等效。
五、结语
枸橼酸西地那非片属临床常用药物,该品种原研产品已在国内上市20余年,临床疗效得到广泛认可,随着该产品国内市场的逐年扩增,近年来成为国内外企业仿制开发的热点品种之一;药物临床试验登记与信息公示平台显示,截至 2022 年 8 月 31日,该品种生物等效试验备案达 50条(含口崩片 17条)。已有生物等效性研究数据可知,枸橼酸西地那非片体内变异总体约 26%,受试者数量一般选择36~48例男性健康受试者开展双交叉、两周期空腹及餐后生物等效性试验;枸橼酸西地那非口崩片则建议参考已发布的《枸橼酸西地那非口崩片生物等效性研究指导原则》仅开展空腹生物等效性试验。同时,建议申请人提交受试制剂与参比制剂的tmax的非参数检验结果,进一步保障仿制药与原研产品的临床等效。




