您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-08-04 16:45
2022 年 4 月 11 日,美国FDA发布《外科缝线 – 基于安全和性能的途径的性能标准》指南文件。旨在提供支持《基于安全和性能的途径》的外科缝线性能标准。计划使用该途径提交外科缝线510(k) 申请的申请人,可以使用该指南中性能标准来支持实质等效性,而不是直接比较目标器械与等价器械的性能。现对该指南内容简述如下:
一、背景
2019 年 9 月,FDA 针对某些已充分了解的器械类型发布了一项旨在描述一种可选途径的指南,即《基于安全和性能的途径》。申请人可以证明新器械符合 FDA 确定的性能标准来证明该器械与合法上市器械一样安全有效。由FDA 确定性能标准,并表示性能满足同类现有合法上市器械要求。
FDA 凭借工作人员的经验和专业知识、文献信息,以及对 FDA 可获得的合法上市外科缝线数据,进行分析来确定性能标准和相关测试方法,以支持本指南中所述外科缝线实质等效性的发现。利用本指南中性能标准提供了一项符合公共健康要求且负担较小的政策。
二、范围/器械描述
本指南适用的外科缝线为符合表 1 所列法规和产品代码的可吸收性和非吸收性缝线。这些器械属于 II 类器械,用于软组织的缝合和/或结扎。
表 1:本指南适用的外科缝线
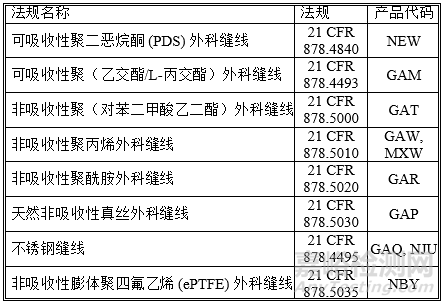
以下外科缝线类型不属于本指南范围:
●重组 DNA 技术生产的可吸收性聚(羟基丁酸)外科缝线 (21 CFR 878.4494, NWJ)
●用于腱索修复或置换的非吸收性膨体聚四氟乙烯外科缝线 (21 CFR 878.5035, PAW)
●可吸收性外科肠线 (21 CFR 878.4830, GAL)
预期用途/使用适应症:
属于本指南文件范围的外科缝线用于一般软组织缝合和/或结扎,例如用于眼科、心血管、神经科、骨科和牙科手术。例如,适用于软组织接合、腹部伤口闭合、疝修补、胸骨闭合和某些骨科手术(包括环扎术和肌腱修补术)的不锈钢缝线都在本指南范围内。
器械设计和其他特征:
非吸收性缝线被描述为多丝或单丝、不可吸收、无菌、柔性、金属、天然或聚合线,用于软组织缝合和结扎、腹部伤口闭合、肠吻合、疝修补和胸骨闭合等用途。可吸收性缝线被描述为多丝或单丝、可吸收、无菌、柔性、合成线,用于软组织缝合和结扎。这两种缝线可含涂层也可不含涂层,可不染色也可经获批颜色添加剂染色(必须符合 21 CFR 70.5(c) 关于缝线使用颜色添加剂的要求),可连接缝针也可不连接缝针。
具有以下特征类型的外科缝线不在本指南范围内:
●由表 1 所列材料以外的材料组成的缝线。
●由动物源性材料组成的缝线。
●包含药物或生物制剂的缝线。
●具有非典型设计特征的缝线 [包括倒钩、环、锚、结、独特的编织图案(例如,包含可膨胀芯的缝合带、编织物)]、配有专用输送工具的缝线 [包括空心针或套管(即非带线缝针)] ,以及具有其他需要性能评估的设计特征的缝线。
●含有尚未获批用于特定缝线材料的颜色添加剂的外科缝线。
●使用 FDA 指南《标记为无菌的器械上市前通告(510(k))提交材料中无菌信息的提交和审查》中所述新型灭菌方法进行灭菌的缝线。
三、测试性能标准
建议提供所有评估试验的结果摘要,以及下文每项试验或评估建议的其他提交信息(例如符合性声明DoC),FDA 可在必要时要求和审查证明新器械符合 FDA 确定性能标准和测试方法的基础数据。所有外科缝线符合现行 FDA 认可的《美国药典》(USP) 版本的专论和章节要求,建议按照“非吸收性缝线专论”或“可吸收性缝线专论”,对无菌缝线成品(例如针刺、卷轴)进行所有测试。除非在下文章节中另有说明,应提交结果摘要、试验方案和完整试验报告等试验信息。
1.试验名称:可吸收性缝线直径(仅适用于可吸收性外科缝线)
方法:FDA 认可的 USP 43-NF38 版本 <861> “缝线 – 直径”
性能标准:10根样品的平均直径以及 30 次测量值中不少于 20 次测量值均在 USP 43-NF38 (2020)“可吸收性外科缝线”表 2 中可吸收性缝线专论针对标签所示尺寸合成缝线规定的平均限值范围内。观察到的单个测量值均不得小于或大于 USP 43-NF38 (2020)“可吸收性外科缝线”表 2 中规定的单个直径限值。
性能标准来源:FDA 认可的 USP 43-NF38 (2020) 版本“可吸收性外科缝线”
不符合性能标准的缝线:如果缝线不符合 USP 的直径要求(例如直径过大),但可能仍然适用于《基于安全和性能的途径》,前提是缝线直径不大于 1 个 USP 尺寸(即给定尺寸的最大直径不超过下一个较大尺寸的最大单个直径限值)。标签应明确说明缝线属于非 USP 缝线,并且应标识每种缝线尺寸的最大超出直径。这些信息可以表格形式呈现,其中标识每种拟定尺寸的 USP 尺寸、相关 USP 直径范围和最大超出尺寸。
提交信息:结果摘要和 DoC
2.试验名称:非吸收性缝线直径(仅适用于非吸收性外科缝线)
方法:FDA 认可的 USP 43-NF38 版本 <861> “缝线 – 直径”
性能标准:10根样品的平均直径以及 30 次测量值中不少于 20 次测量值均在 USP 43-NF38 (2020)“非吸收性外科缝线”表 1 中针对标签所示尺寸规定的平均限值范围内。观察到的单个测量值均不得小于或大于 USP 43-NF38 (2020)“非吸收性外科缝线”表 1 中规定的单个直径限值。
性能标准来源:FDA 认可的 USP 43-NF38 (2020) 版本“非吸收性外科缝线”
不符合性能标准的缝线:如果缝线不符合 USP 的直径要求(例如直径过大),但可能仍然适用于《基于安全和性能的途径》,前提是缝线直径不大于 1 个 USP 尺寸(即给定尺寸的最大直径不超过下一个较大尺寸的最大单个直径限值)。标签应明确说明缝线属于非 USP 缝线,并且应标识每种缝线尺寸的最大超出直径。这些信息可以表格形式呈现,其中标识每种拟定尺寸的 USP 尺寸、平均直径的相关限值和最大超出尺寸。
提交信息:结果摘要和 DoC
3.试验名称:针线连接强力
方法:FDA 认可的 USP 43-NF38 版本 <871> “缝线 – 针线连接强力”
性能标准:5 个值的平均值或任何单个值均不小于 USP 43-NF38 <871> “缝线 – 针线连接强力”表 1 中指定尺寸的限值。
性能标准来源:FDA 认可的 USP 43-NF38 (2020) 版本<871> “缝线 – 针线连接强力”
提交信息:结果摘要和 DoC
4.试验名称:可吸收性缝线断裂强力(仅适用于可吸收性外科缝线)
方法:FDA 认可的 USP 43-NF38 版本 <881> “缝线 –断裂强力”
性能标准:平均断裂强力不低于 USP 43-NF38 (2020) “可吸收性外科缝线”表 2 中可吸收性缝线专论针对标签所示类别和尺寸合成缝线规定的平均断裂强力。
性能标准来源:FDA 认可的 USP 43-NF38 (2020) 版本“可吸收性外科缝线”
提交信息:结果摘要和 DoC
5.试验名称:非吸收性缝线断裂强力(仅适用于非吸收性外科缝线)
方法:FDA 认可的 USP 43-NF38 版本 <881> “缝线 – 抗张强度”
性能标准:平均断裂强力不低于 USP 43-NF38 (2020) “非吸收性外科缝线”表 1 中针对标签所示类别和尺寸规定的平均断裂强力。
性能标准来源:FDA 认可的 USP 43-NF38 (2020) 版本“非吸收性外科缝线”
提交信息:结果摘要和 DoC
6.试验名称:可吸收特性(仅适用于可吸收性外科缝线)
方法:可吸收特征测试应包括记录器械的吸收速率、至完全吸收时间、随时间变化的残留断裂强力。可吸收特征应以图表、表格或图形形式呈现,以说明缝线在具有临床意义的时间段内的残留断裂强力。具有临床意义的时间长度取决于缝线的预期用途。建议使用可适当复制外科缝线预期使用方式、部位的模型进行体内研究。
应对大量缝线进行测试,以证明断裂强力保持率一致。测试应至少包括缝线最大、最小尺寸以及两者之间尺寸,所测试尺寸之间跳过的尺寸差异不得超过两个尺寸。例如,如果拟上市 7 到 7-0 范围内的所有缝线尺寸,建议测试尺寸 7、4、1、2-0、5-0 和 7-0 的断裂强力保持率。性能标准:器械的可吸收特征与器械的预期用途一致(例如,软组织的短期或长期接合)。
性能标准来源:外科缝线 – 行业及 FDA 工作人员的 II 类特殊控制指南
提交信息:结果摘要和测试方案
7.试验名称:灭菌(适用于标记为无菌的器械)
方法:以下共识标准的现行 FDA 认可版本(如适用):
ISO 17665-1 医疗保健产品灭菌 – 湿热 – 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制要求
ISO 11135-1 医疗保健产品灭菌 – 环氧乙烷 – 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制要求
ISO 11137-1 医疗保健产品灭菌 – 辐射 – 第 1 部分:医疗器械灭菌过程的开发、确认和常规控制要求
ISO 20857 医疗保健产品灭菌 – 干热 – 医疗器械灭菌过程的开发、确认和常规控制要求
ISO 11607-1 最终灭菌医疗器械的包装 – 第 1 部分:材料、无菌屏障系统和包装系统的要求
ISO 11607-2 最终灭菌医疗器械的包装 – 第 2 部分:成形、密封和装配过程的确认要求
性能标准:确认测试应证明器械的无菌性或灭菌能力达到 10-6 无菌保证水平。
性能标准来源:FDA 指南:《标记为无菌的器械上市前通告(510(k))提交材料中无菌信息的提交和审查》
提交信息:应提供包装(无菌屏障系统)描述及其如何保持器械无菌性,以及包装测试方法描述。无论是使用何种灭菌方法,都应提供 FDA 指南《标记为无菌的器械上市前通告(510(k))提交材料中无菌信息的提交和审查》第 V.A. 节中的信息。
8.试验名称:货架有效期
方法:应使用经老化的缝线遵循上述第 1-7 项试验(如适用)中列出的性能测试方法。应根据拟定货架有效期对至少三个生产批次的老化器械进行测试。
性能标准:为了证明在标示货架有效期内的持续无菌性、包装完整性和器械功能性,经老化的缝线应符合上述第 1-7 项试验(如适用)中列出的相应性能标准。
性能标准来源:请参见上述第 1-7 项试验的性能标准来源。
其他考虑事项:如果使用已加速老化的器械进行货架期试验,建议具体说明器械老化的方式。根据 ASTM F1980《医疗器械无菌屏障系统加速老化的标准指南》现行版本对器械进行老化处理,并指定为达到失效日期而确定的环境参数。对于含有聚合材料的缝线,应计划对实时老化样品进行测试,以确认加速老化可反映实时老化。
建议提供用于货架有效期试验的试验方法、结果以及结论的摘要。
生物相容性评价
使用 FDA 指南《使用国际标准 ISO 10993-1》“医疗器械的生物学评价 - 第 1 部分:风险管理过程中的评价与试验”的附件 A,在本文其余部分简称为《FDA 生物相容性指南》。外科缝线的接触类型、持续时间取决于预期用途,将其视为与组织/骨骼以及血液(在某些情况下)长时间或持久接触的植入器械,应在生物相容性评价中说明以下终点。
●细胞毒性
●致敏性
●刺激性或皮内反应性
●急性全身毒性
●材料介导的致热原性
●亚急性/亚慢性毒性
●遗传毒性
●临床相关性植入
●慢性毒性
●致癌性
与血液长时间或永久接触的缝线的其他终点包括:
●血液相容性
代替测试的理由:如果目标器械是使用相同的组织接触类型、持续时间的等价器械的原材料、制造工艺,并且几何结构的任何变化预期不会影响生物反应,这通常足以确定实质等效生物相容性。
测试:如《FDA 生物相容性指南》附件 E 所述,如果确定需要进行测试来说明部分或所有已确定终点, 建议提供所有执行试验的完整试验报告,除非可以适当提供无补充信息的符合性声明。任何试验特定的阳性、阴性和/或试剂对照样品应符合预期,应充分描述和证明方案偏离。如果器械无法完全凭借 FDA 确定的性能标准来证明其提交材料具有实质等效性,则不适用于《基于安全和性能的途径》计划;但是,之前确定的针对相应等价器械进行直接性能比较的 510(k) 计划(包括传统、特殊和简略 510(k) 计划)仍然可用。
9.试验名称:生物相容性终点(根据《FDA 生物相容性指南》确定)
方法:生物相容性共识标准的现行 FDA 认可版本
性能标准:应确定与组织直接接触的所有器械组件都具有可接受的生物反应
性能标准来源:《FDA 生物相容性指南》
其他考虑事项:对于任何具有不良生物反应的生物相容性试验样品,生物相容性评价应说明所观察到的毒性水平可接受的原因。可能需要针对合法上市的等价器械进行一些比较测试,以支持《FDA 生物相容性指南》中说明的理由。对于标准生物相容性试验方法(包括对比器械对照样品),合法上市对比器械的对照样品应符合预期。
对于配备缝针的外科缝线,应证明符合 FDA 认可的标准(例如,针对不锈钢),并提供理由说明为何无需对缝针进行生物相容性试验。如果无法证明符合公认的标准和/或如果缝针含有涂层,则应根据 ISO 10993-1 对缝针组件进行生物相容性试验以确定相应接触分类,例如接触时间有限的外部连通器械(< 24 小时)。
以上是对FDA该文件主要内容的简介。对其进行研究探讨,有助于借鉴国际先进经验和工作方式,提高我国对外科缝线产品的审评效率,从而更好地促进我国医疗器械行业的发展。

来源:中国器审


