您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-05-12 04:04
一、MDR法规简介
2017年5月5日,欧盟官方期刊(Official Journal of the European Union)正式发布了欧盟关于医疗器械第2017/745号法规(简称“MDR法规”)。MDR法规于2017年5月26日正式生效,并于2021年5月26日正式取代MDD(93/42/EEC)和AIMDD(90/385/EEC)。
MDR共10章123条,17个附录。相较于MDD,在内容上,法规在整合原指令的基础上,大幅提升了有关医疗器械认证的规范和限制,例如关于产品分类规则、器械的可追溯性、临床性能研究的规范、增加上市后的产品安全性和有效性的监管等方面。在监管级别上,MDR由指令升级为法规,这是欧盟医疗器械管理法规发生的重大变化,提高了对欧盟成员国的约束力,无需各国转化为本国的法律法规的形式即可落实实施。MDR法规在监管层面上的变化主要包括三个方面:
1.提高医疗器械的质量、安全性和可靠性
新法规要求对植入产品等高风险器械实施更严格的监管,某些高风险设备(例如植入物)可能需要欧盟层面的独立专家小组审核。还将加强对临床试验以及经授权的公告机构的监管。新法规还将涵盖某些以前不受监管的美容产品(例如,没有矫正视力作用的彩色隐形眼镜)。
2.增强产品的透明度
新法规引入欧洲医疗器械数据库(EUDAMED),为产品运营商,患者和公众提供欧盟市场上可用医疗器械信息。新法规将确保重要信息易于查找。例如,将为患者引入带有所有植入物相关重要信息的植入卡,并且每个产品都必须有唯一的器械标识符(UDI),以便在整个供应链中的可追溯性,这将确保出现问题时能够迅速采取措施。
3.加强警戒和市场监控
一旦器械进入欧盟市场,制造商将有义务收集有关其性能的数据,欧盟成员国有义务鼓励医疗专业人员,用户和患者使用标准化格式在国家层面报告可疑事件。
二、MDR法规下医疗器械产品分类及监管主体
MDR法规将医疗器械分成Ⅰ、Ⅱa、Ⅱb和Ⅲ类四个类别,详见表1。
表1 MDR法规下医疗器械分类及监管主体

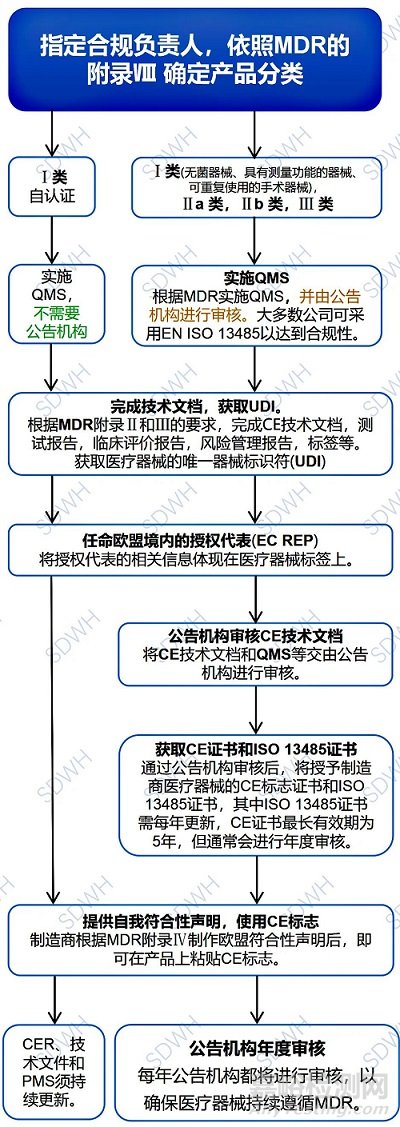
三、 MDR法规下CE标志申请流程
CE标志是欧盟认可医疗器械安全性和质量的重要标志,是医疗器械产品进入欧盟市场的前提。欧盟对医疗器械产品进行分类管理,不同类目下的医疗器械,其认证所需的审核程序不同。MDR法规对各类医疗器械的CE标志申请程序提出了新的要求,具体申请流程和各环节所需的关键资料如下图所示。
参考资料
【1】林晓君. 欧盟MDR法规下医疗器械产品监管机制解读[J]. 大众标准化,2020(13):146-148.
【2】https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0745
【3】https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0745

来源:苏大卫环


