您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-02-12 22:20
编者语
Mzwinsunny曾发表过《溶出试验的作用原来如此强大,且看FDA观点!》译文,原英文题目为Dissolution Testing for Generic Drugs: An FDA Perspective。文中有一节对调释制剂乙醇诱导的剂量倾泻做了简要介绍,包括做剂量倾泻试验的原因、FDA对申报品种的要求、体外剂量倾泻试验的溶出条件等。
CFDA在2016年3月18日发布了公告(2016年第61号),其附件《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》中规定:饮用含酒精的饮料可能会影响药物自调释制剂中释放。酒精会改变药物释放特性,导致药物过快释放,并改变药物体内暴露量,进而影响药物的安全性和有效性。建议研发缓释口服固体制剂时进行相应的体外研究,以评价制剂在体内酒精环境中出现药物突释的可能性。应考察制剂在不同浓度的酒精溶媒中的释放情况。某些特定情况下可能需要进行制剂与酒同服时的生物等效性研究。”这个指导原则紧跟国外研究情况,填补了国内多年的空白,对调释制剂仿制药研发、注册申报、一致性评价具有重要意义。
为了使业内同行进一步了解剂量倾泻,小编对国外相关资料进行了翻译整理,希望对各位有所帮助,同时欢迎各位专家、同行对相关指南、技术要求、制剂处方工艺开发进行探讨。
原文标题
Regulatory Considerations for Alcohol-Induced Dose Dumping of Oral Modified-Release Formulations
Thomas P. Friebe, Firouz Asgarzadeh, Ann Gray, Kevin Hughes, Johann-Philipp Hebestreit, Yvonne Rosiaux, Mahmud Yunis, Amina Faham
在过去几十年里,调释(MR)系统已经彻底改变了原料药的给药方式。通过保持更一致的血药浓度,患者的依从性得到了改善,副作用得到了减少,促进了更有效的治疗。绝大多数患者受益于调释制剂的药理优势和服用的方便性。然而,当含乙醇的饮料与调释药物同服时,有一部分患者会产生意外的药物过量。由于药物从调释系统中的释放是由聚合物基质或由聚合物包衣控制的,如果制剂在含乙醇的液体中溶解,释放控制就会遭到破坏,就可能会发生剂量倾泻(Dose Dumping)。
“剂量倾泻”是指药物在短时间内全部或绝大部分快速释放(1)。基于API的治疗指数、药代动力学特性和适应症,可能会导致严重的副作用甚至致命。药物与含乙醇饮料同服而引起的剂量倾泻,称为“乙醇诱导的剂量倾泻”(ADD)。
调释释放——有益,也有风险
乙醇饮料消费遍及全世界各地。某些病人,如患有慢性疼痛或抑郁症的人,有可能倾向于采用饮酒作为一种处理方式来应对他们的疾病,这是因为酒的生理效应与麻醉剂类似(2)。尽管产品说明书中做出了有关警告,但是如果患者将药品与乙醇饮料同时服用,就可能导致意外的ADD发生。虽然二者很少同时服用,但是由于残留的乙醇存在,ADD仍然可能会发生。如果一个人故意使用高浓度的乙醇饮料从缓释制剂中提取高剂量的API(通常是阿片类镇痛药),那么故意的ADD就会发生。
本文不去关注这种故意的ADD(包括防滥用处方),而是强调调释制剂应具有一个恰当的、耐受的处方来确保病人的安全,例如,对于窄治疗窗药物。
2005 年的“Palladone事件”使监管部门提高了对ADD的认识(3,4)。Palladone是一个氢可酮缓释胶囊,采用甲基丙烯酸共聚物B型和乙基纤维素(5)作为控制药物释放的材料,二者都溶于乙醇。在健康受试者中进行的药代动力学研究表明,共同服用240ml(8盎司)40%(80度)的乙醇饮料和12mg的palladone胶囊后,与用水服用相比,会导致氢吗啡酮的平均血药浓度提高6倍。这么高的浓度可能是致命的(6)。因此,Palladone从美国市场撤市了。这一事件随后催生了新的指南,要求企业在进行制剂处方开发过程中必须要考虑ADD。
监管机构的考虑
欧盟(7-9),美国(1,10)和其他国家(11)的监管机构已经提出了关于ADD的指南,然而ICH却还没有提出。迄今为止,还没有一个监管机构将所有ADD相关要求汇总在同一份文件中。
欧盟 根据指南(8)和EMA质量工作组(QWP)(9)Q&A部分要求,应进行乙醇存在下的体外试验。申请人需要评估所有类型调释制剂处方中API非预期释放的风险,如果出现了ADD或可能出现ADD,产品应该重新设计处方。图1提供了决策图。
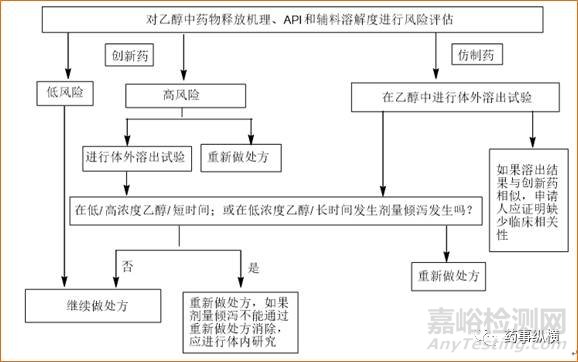
图1. 基于EMA指南(8,9)的乙醇诱导剂量倾泻(ADD)风险评估
2011年,一份EMA评估报告(12)要求上市许可持有人(MAH)提供他们阿片类产品对乙醇敏感性的数据。共有8个申请人提交了14个产品的数据。然而,每个申请人采用了不同浓度的乙醇进行评价。在这种情况下,不但方法不够有效,而且使审评人员很难进行判断和数据比较。最近EMA公布的Q&A(9)中提供了所需浓度乙醇的具体指南。
美国。2011年,FDA仿制药办公室(OGD)对仿制药提供了一个简单的流程(13,14)来应对ADD,见图2。
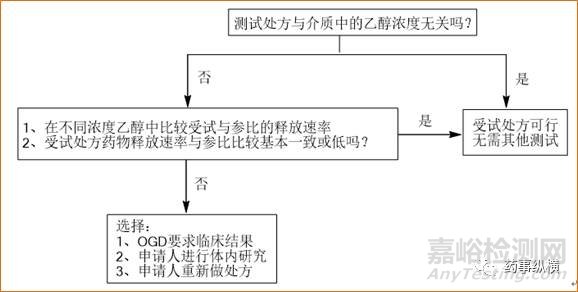
图2.FDA乙醇诱导的剂量倾泻(ADD)应对流程-关键仿制药处方(13)
2014年以来,有一份工业指南(15)规定,会把不充分的溶出数据作为理由来拒收ANDA申请。此外,FDA对于一些API提供了详细的生物等效性溶出建议(16),如非阿片类药物琥珀酸美托洛尔(17)、可乐定(18)、美金刚(19),二甲双胍和西他列汀磷酸(20),曲司氯铵(21),这些是广泛使用的治疗慢性疾病的药物。通常对于这些药物,乙醇测试浓度需要达到40%。
FDA和EMA要求比较
EMA和FDA关于ADD的要求并非完全一致,有时甚至相互矛盾。表1显示了它们之间的差异主要包括三方面。FDA要求溶出试验中采用40%乙醇作为介质,而EMA要求20%。应当说明的是,胃中如果达到40%的乙醇浓度需要摄入240ml含56%乙醇的饮料(基于已有100ml胃液存在于胃中)(4,22)。
这么大的乙醇摄入量在极端情况下似乎是可以实现的,即“酗酒”。此外,乙醇通常在30分钟内在胃肠道迅速吸收和消除(23)。因此,在体外要求对40%乙醇耐受似乎更与乙醇滥用相关,而20%乙醇可能更接近出现意外ADD的情况。
EMA和FDA要求之间的差异可能会使处方设计师会产生困惑,不知该遵循哪个。由于许多制药公司在全球范围内经营,他们不希望在不同地区卖不同处方的产品,因此可能强制要求处方设计师开发出对40%乙醇耐受的处方,不管这是否与生理相关。这对处方开发是一个明显的技术障碍,甚至可能阻碍有价值的药物的推出。

乙醇敏感性评估
原料药和/或辅料对乙醇敏感并不一定意味着制剂会发生剂量倾泻。处方设计师需要评估这一敏感程度,而这样做时,处方设计师需要采用已被普遍接受的工具或指南。N. Jedinger等人(25)就如何应对这个问题讨论了一些有意义的方法来减少ADD的风险,包括:影响ADD的理化因素、恰当的基质系统和技术策略。
通过比较每个溶出取样点的相似性可以评价溶出行为的相似性。有不同方法可以进行溶出行为比较,但是一个在FDA关于NDA和ANDA的文件里(26-28)讨论最多的如下:
模型非依赖法是使用差异因子(f1)和相似因子(f2)来比较溶出行为(29)。差异因子(f1)用来计算两条曲线每个时间点的溶出百分比的差异。相似因子(f2)用来衡量两条曲线溶出百分比的相似性。如果两条曲线相似,f1值应接近0、f2值应接近100。通常,f1值达到15(0-15)、f2值大于50(50-100)说明两条曲线相似。
复杂因素
文中前部分所描述的法规指南是关于体外测试的,这个要求是有必要的,因为如果在临床试验中采用志愿者来测定ADD的风险可能会引起不必要的风险,这被视为是不道德的。处方设计师把一个复杂的事件简单化后会导致过于强调某些因素(如纯聚合物在乙醇的溶解性)而忽视了其他因素(如处方设计)。
成功开发一个处方需要考虑药物和辅料的性能以及处方的设计。事实上,监管部门要求的40%乙醇介质的体外结果不一定能够预测体内行为,体内环境的复杂性使其缺少相关性,以下两个例子就可以很好说明:
●通常认为缓释骨架片是耐ADD的。Opana是一个羟吗啡酮缓释片,为亲水性聚合物处方(TIMERX药物递送技术),采用的调释聚合物——黄原胶(30)和刺槐豆胶(31)都是醇不溶性胶体。Opana虽然通过了体外ADD溶出试验,但在体内却是失败的(32)。
●尽管多颗粒制剂通过胃的转运时间比单片要短,但是经包衣调释多元制剂通常认为会产生ADD,因为其具有较高的有效接触表面使酸性水醇介质进入。卡维地洛是一个多颗粒制剂(微型泵),采用甲基丙烯酸共聚物作为包衣材料。甲基丙烯酸共聚物通常在乙醇中溶解,在体外溶出试验中没有通过ADD。然而,其体内特性却不受乙醇的影响(33)。
上面这两个例子说明,体内的ADD是一个多因素事件。因此,当前所采用的标准体外评价方法不一定能够预测体内行为。显然,这需要一个经过特殊设计的测试方法,必须根据具体情况去选择恰当的处方和工艺,要考虑多种因素,如:原辅料性质、处方设计、适应症、剂量倾泻的风险。
总结
调释制剂的ADD对于一部分患者可能具有风险。监管部门已经建立了指南来指导处方设计师降低处方原因所引起的ADD。然而,仅仅进行体外测试是不能够代表生理环境的,这可能在以合理的成本开发有效剂型方面造成技术障碍。当前,不同地区间缺少统一的要求,这增加了研究开发的复杂性,增加了全球化企业的成本。鉴于制药行业的日益全球化,FDA和EMA指南应该协调有关ADD的体外试验条件,例如能反映生理相关的乙醇浓度和暴露的时间。
国际要用辅料协会(IPEC)欧洲ADD工作组
国际药用辅料协会(IPEC)欧洲ADD工作组计划公布一份意见书强调ADD的有关问题,并建议对当前的指南进行修订,以为配方设计师提供相关的建议。

来源:药事纵横


